

Copyrights: Bui The Dung, Tran-Thi Xuan Anh, Tran Hoa, Nguyen Minh Duc, 2023. License: This work is licensed under a Creative Commons Attribution 4.0 International License.
Abstract
This case report highlights the successful management of a patient with light chain amyloidosis and New York Heart Association (NYHA) functional class IV heart failure who was ineligible for an autologous stem cell transplant (ASCT). They were managed with an oral melphalan and dexamethasone regimen in addition to standard heart failure treatment according to European Society of Cardiology-American Heart Association guidelines. They responded well to treatment, showing symptom improvement and the ability to function at the NYHA class II level. They were closely monitored throughout treatment, with regular testing for infective screening, systemic biochemical workup, echocardiography, and monitoring of N-terminal pro-B-type natriuretic peptide (NTproBNP) and troponin T levels. After 24 cycles of high-dose oral melphalan and dexamethasone chemotherapy, the patient could discontinue treatment and continue follow-up monitoring. The most recent workup showed significantly improved cardiac function, with decreased left ventricular wall thickness, improved diastolic function and strain, and decreased NT-proBNP level. This case report demonstrates the importance of considering alternative treatment options for patients ineligible for ASCT due to advanced heart involvement. It also highlights the potential benefits of an oral melphalan and dexamethasone regimen combined with standard heart failure treatment in improving clinical outcomes in these patients.
Introduction
Light chain amyloidosis (AL) is a rapidly progressing type of cardiac amyloidosis (CA)1, and prompt initiation of therapy can significantly affect patient outcomes. Diagnosing CA requires an accurate and comprehensive diagnostic workup to identify the affected organs and assess the severity of organ involvement2. The treatment of AL is risk-adapted, with considerations for clonal characteristics, comorbidities, and the severity of organ involvement. Therapy aims to deliver a rapid, effective, and well-tolerated treatment. While autologous stem cell transplant (ASCT) is the first choice, the response to chemotherapy is modest and can vary depending on the formula selected. Despite advances in treatment, CA remains a severe and progressive condition3.
Limited data on CA in Viet Nam makes documenting successful treatment outcomes and survival rates important, as we do in this case report. By sharing such cases, we hope to improve our understanding of CA and identify opportunities for its better diagnosis and management. These efforts can help improve patient outcomes.
We present a case report of a 38-year-old man with worsening dyspnea and orthopnea diagnosed with CA who received prompt diagnosis and treatment.






CASE REPORT
A 38-year-old man with an unremarkable medical history was admitted to the Cardiology Department with worsening dyspnea and orthopnea. The patient had a systolic blood pressure of 90 mmHg at admission, with no orthostatic hypotension. Physical examination revealed moderate to large bilateral pulmonary effusion, elevated jugular venous pressure, and mild lower extremity edema.
The patient’s electrocardiogram (ECG) showed sinus rhythm with low QRS voltages in the anterior and inferior leads and Q-waves in V1-V2-V3, which were inconsistent with the degree of left ventricular (LV) hypertrophy (Figure 1). Echocardiography showed a hypertrophic LV with an LV septal diameter of 23 mm, posterior wall diameter of 22 mm, hypokinetic wall motion, and preserved ejection fraction (53%; Figure 2 A). The patient showed grade III diastolic dysfunction (E/A > 1.5–2.1; E/e’ = 18–20; Figure 2 B-C), reduced LV global longitudinal strain (−14%) with an “apical sparing” pattern (Figure 2 D), thickened mitral valve leaflets, LV hypertrophy with sparkling signs, and a dilated inferior vena cava.
Cardiac magnetic resonance imaging (MRI) revealed diffuse ventricular wall thickening with an LV thickness of 15–19 mm (LV septum thickness = 19 mm, anterior wall thickness = 16 mm, lateral wall thickness = 15 mm, and inferior wall thickness = 16 mm) and a right ventricular wall thickness of 9–10 mm in the diastolic phase. No LV outflow obstruction was observed. The patient had mild dilation of the atria and mild thickening of the atrial septum. In the late gadolinium enhancement (LGE) phase, a moderate-to-strong signal increase was observed in the mid-wall and endocardium of the right ventricle and LV walls from the basal area to the apex and atrial septum (Figure 3). Additionally, the time of inversion sequence was indeterminate. The laboratory workup revealed high levels of N-terminal pro-B-type natriuretic peptide (NT-proBNP; 22,011 pg/mL) and troponin T (TropT; 0.141 ng/mL).
Based on the patient’s clinical presentation and workup, the suspected diagnosis was cardiomyopathy with hypertrophy and a restrictive pattern. Serum immunofixation was performed, and urine immunofixation was unavailable. Kappa and lambda light chains were not detected in the initial immunofixation assay. However, a second assay showed an abnormal free kappa-lambda ratio (free kappa = 385 mg/mL, free lambda = 217 mg/dL, ratio = 1.77). An abdominal fat biopsy was examined with Congo red staining and electron microscopy. It showed amyloid deposits in the vessel walls that were positive for Congo red stain, and apple-green birefringence appeared under polarized light. Therefore, secondary amyloidosis could be excluded (Figure 4).
TREATMENT
The patient was diagnosed with advanced cardiac involvement and classified as Mayo cardiac stage III with AL and New York Heart Association (NYHA) functional class IV heart failure. ASCT was not a viable treatment option due to the patient’s frailty and contraindication to transplant with a high NYHA functional class (III or IV). Therefore, they were treated with oral melphalan (10 mg/m2 of the body surface area) and high-dose intravenous dexamethasone (40 mg/day) on days 1–4 for up to 24 cycles (MDex chemotherapy) in addition to standard heart failure treatment according to the European Society of Cardiology-American Heart Association guidelines.
Before initiating treatment, the patient underwent an extensive infectious disease workup, including screening for cytomegalovirus, Epstein–Barr virus, herpes simplex virus (immunoglobulin G and A), hepatitis B virus (surface and e antigens), hepatitis C virus, tuberculosis, C-reactive protein, and procalcitonin; a chest X-ray; and a comprehensive biochemical evaluation. Echocardiography, NT-proBNP, and TropT were used to monitor treatment response.
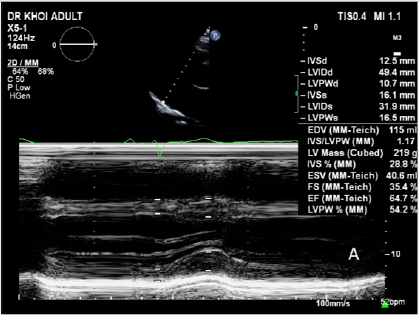
After completing 24 cycles of MDex chemotherapy, the patient’s symptoms improved, and their NYHA functional class improved from IV to II, enabling them to resume work. The patient has remained stable for approximately five years, with no hospitalizations and a sustained NYHA functional class of II. They undergo monthly outpatient clinic visits for follow-up and monitoring for any signs of relapse. A recent workup showed a significant improvement in diastolic function and strain and decreased LV wall thickness (Figure 5). Their NT-proBNP level was 1645 ng/mL, which decreased markedly during chemotherapy (Figure 6). Their serum kappa was 470.1 mg/dL (reference range: 156–408), and their serum lambda was 280.7 mg/dL (reference range: 83–224), with negative immunofixation electrophoresis.
DISCUSSION
The prevalence of amyloidosis, a rare but serious medical condition, is increasing globally, but with limited data available in Asia, particularly in Vietnam. From July 2017 to March 2023, 21 amyloidosis cases were diagnosed at Ho Chi Minh University of Medicine and Pharmacy Hospital using clinical signs and imaging techniques such as echocardiography and MRI. The definitive diagnosis was confirmed by histological pathology and abnormal monoclonals in the blood. AL accounted for most cases (90%), and the patients’ mean age was 63 years. Access to specific treatment was limited, with only five cases receiving treatment, and mortality during the six-month follow-up period was 50%. Additionally, almost half of the patients experienced arrhythmia, with a relatively similar occurrence of atrial arrhythmia, severe ventricular arrhythmia, bradyarrhythmia, and atrioventricular conduction block. These findings highlight the importance of further research into the prevalence and treatment of amyloidosis in Asia, particularly in Viet Nam, to improve the understanding and management of this serious medical condition.
Amyloidosis is the abnormal deposition of an extracellular protein called amyloid, which can result from abnormally functioning plasma cells, mutations in a hepatocyte protein called transthyretin, or protein A formation due to systemic inflammatory diseases. The chemical composition of the deposited amyloid protein is used for modern classification, which guides targeted treatments. The disease prognosis depends on the number of organs involved and the extent of infiltration. Various classification methods have been developed. However, the modern classification based on the chemical composition of the deposited amyloid protein has recently been widely used since it provides a more direct orientation for target treatments that were well-described in 20214. AL is the most common form of amyloidosis and can affect multiple organs, including the kidneys, liver, nerves, and heart. Its prognosis depends on the number of organs involved and the extent of infiltration. Multi-modal diagnostic imaging can help detect amyloidosis at an early stage. However, despite advancements in treatment, mortality and morbidity rates remain high for CA.
Immunofixation showing a monoclonal plasma cell disorder is required to establish a systemic AL diagnosis with high sensitivity (>95%)5. However, it can be negative in 30% of patients with AL using serum protein and immunofixation electrophoresis because they typically have low monoclonal protein amounts6. Our patient’s first immunofixation assay was negative, but the following workup showed an abnormal free kappa-lambda ratio (1.77).
Echocardiography is the initial imaging test for CA. Our patient’s ECG met all these features with significant LV wall thickness, which was confirmed by a cardiac MRI. The myocardium and other heart structures, such as atria, valve leaflets, and atrial septum. LV wall thickening was incompatible with the patient’s twelve-lead ECG, which showed no criteria for LV hypertrophy.
While echocardiography is the preferred imaging method for identifying CA, until recently, it has been challenging to differentiate CA from other diseases. Myocardial strain imaging is a contemporary echocardiographic technique with more advantages and provides sensitive and specific indicators for the presence of CA. Strain echo shows an apical sparing pattern signature with good sensitivity and specificity for differentiating CA from other LV hypertrophy diseases. According to the 2016 American Society of Echocardiography/European Association of Cardiovascular Imaging Recommendations, grade III diastolic function was associated with reduced LV global longitudinal strain7.
The simplest and most common adjunctive diagnostic tool for CA is ECG. The most common ECG findings are a typical low voltage on limb leads and a pseudo-infarct pattern, present in 54.5% and 40.2% of patients with CA and 20.7% and 4.6% of patients with non-cardiac involved amyloidosis, respectively8. Our patient’s ECG showed a sinus rhythm, left axis due to left anterior hemiblock without disturbance in depolarization, and low QRS voltages in the limb and precordial leads.
Cardiovascular MRI is also a key modality in diagnosing CA. It provides detailed information on cardiac structure and function and myocardial tissue characteristics. The degree of amyloidosis infiltration correlates with LGE progression (none, subendocardial, or transmural). Our patient’s cardiac MRI showed diffuse LV thickening with wall thickening, right ventricular wall thickening, and subendocardial LGE indicative of amyloid infiltration.
Congo red-stained amyloid deposits in endomyocardial biopsies are the gold standard. However, this procedure is invasive, requires an expert center to be performed, and is associated with greater risk; the biopsy of a surrogate site is preferred. The histologic diagnosis of AL is based on a fat-pad biopsy. This procedure is less invasive and easier to access than a myocardium biopsy. Abdominal subcutaneous fat aspiration was as sensitive as rectal biopsy and substantially more sensitive than bone marrow biopsy in diagnosing amyloidosis (sensitivity = 72%, specificity = 99%)9. A large-bore needle and multi-site aspiration were used in our patient to decrease the likelihood of a false negative result. The Congo red dye showed amyloid deposits in red and a birefringence apple-green appearance under cross-polarized light. Mass spectrometry showed lambda and kappa deposits.
Patients with CA and heart failure have an overall median survival of only six months without disease-specific treatment. The survival of patients with AL with cardiac involvement has significantly improved with modern therapies. Besides heart failure and comorbidity management, such as heart failure and arrhythmia (including atrial fibrillation and heart rhythm conductive disturbance), amyloidosis treatment aims to control the underlying protein misfolding disorder. Chemotherapy and/or ASCT target amyloids produced by plasma cell clones10. According to current AL treatment guidelines, ASCT must be considered before choosing a chemotherapy formula, and bortezomib-based induction therapy before stem cell mobilization and transplantation has a better outcome. Bortezomib-based regimens are also first-line therapy for most patients unsuitable for hematopoietic stem cell transplantation under the current amyloidosis treatment recommendation11, 12. Our patient was unsuitable for ASCT based on common eligibility criteria. Patients classified as NYHA functional class IV or with a large pleural effusion or high TropT level are considered unsuitable for hematopoietic cell transplantation.
Bortezomib-based regimens, including daratumumab with bortezomib and dexamethasone, have increased good partial or complete response and overall survival rates in patients with AL13. In 2018, there were disparities in access to recommended therapies for AL, and oral melphalan with dexamethasone (MDex) was considered a standard of care13, 14. Some chemotherapies were unavailable at our center, so we opted for the MDex regimen with oral melphalan (10 mg/m2 of the body surface area [0.22 mg/m2]) and high-dose intravenous dexamethasone (40 mg/day) for four consecutive days, given every 28 days. Patients with stage III AL cardiac involvement have a poor prognosis, with a five-year survival rate2. The median survival was 4.3 years for those with stage III and one year for those with stage IIIb14, 15. While our patient was ineligible for ASCT, they achieved organ response with conventional regimen chemotherapy. Their mean interventricular septal thickness decreased by 2 mm, ejection fraction improved, NYHA class improved by two without an increase in diuretic use or wall thickness, and NT-proBNP level decreased by >30%. They developed a stable hematologic response and remission for more than five years.
CONCLUSION
AL is a rare disease that can significantly impact patient survival. Nearly 20% of patients newly diagnosed with AL present with advanced cardiac involvement (stage IIIb), which is associated with poor median survival. Early diagnosis is crucial since it allows for prompt immunofixation and differentiation of the amyloidosis type. In addition to clinical workups, imaging is an accessible and essential diagnostic modality for identifying CA. Patients with AL have shown improved response and survival rates with bortezomib-based regimens, particularly when they include daratumumab. Triple therapy with bortezomib, melphalan, and dexamethasone is recommended for patients ineligible for stem cell transplantation over melphalan with dexamethasone. However, this case report indicates a favorable response to initial treatment with oral melphalan and high-dose dexamethasone in a patient with light-chain CA at our center. The case presented in this report showed a sustained response to oral melphalan and high-dose dexamethasone combined therapy for light-chain CA, which is encouraging for the initial management of CA.
Abbreviations
AL: Light chain amyloidosis, ASCT: Autologous stem cell transplant, CA: Cardiac amyloidosis, ECG: Electrocardiogram, NTproBNP: N-terminal pro-B-type natriuretic peptide, NYHA: New York Heart Association
Acknowledgments
None.
Author’s contributions
Bui The Dung and Tran-Thi Xuan Anh: Case file retrieval and case summary preparation. Bui The Dung and Nguyen Minh Duc: preparation of manuscript and editing. All authors read and approved the final manuscript.
Funding
None.
Availability of data and materials
Data and materials used and/or analyzed during the current study are available from the corresponding author on reasonable request.
Ethics approval and consent to participate
University Medical Center HCMC does not require ethical approval for reporting individual cases or case series. Written informed consent was obtained from the patient(s) for their anonymized information to be published in this article.
Consent for publication
Written informed consent was obtained from the patient for publication of this case report and any accompanying images. A copy of the written consent is available for review by the Editor-in-Chief of this journal.
Competing interests
The authors declare that they have no competing interests.
References
-
Wechalekar
A.D.,
Gillmore
J.D.,
Hawkins
P.N.,
Systemic amyloidosis. Lancet.
2016;
387
(10038)
:
2641-54
.
View Article PubMed Google Scholar -
Dispenzieri
A. B.F.,
Kumar
S.K.,
Reeder
C.B.,
Sher
T.,
Lacy
M.Q.,
Kyle
R.A.,
Mikhael
J.R.,
Roy
V.,
Leung
N.,
Grogan
M.,
Kapoor
P.,
Lust
J.A.,
Dingli
D.,
Go
R.S.,
Hwa
Y.L.,
Hayman
S.R.,
Fonseca
R.,
Ailawadhi
S.,
Bergsagel
P.L.,
Chanan-Khan
A.,
Rajkumar
S.V.,
Russell
S.J.,
Stewart
K.,
Zeldenrust
S.R.,
Gertz
M.A.,
Treatment of Immunoglobulin Light Chain Amyloidosis: Mayo Stratification of Myeloma and Risk-Adapted Therapy (mSMART) Consensus Statement. Mayo Clinic Proceedings.
2015;
90
(8)
:
1054-1081
.
View Article Google Scholar -
Muchtar
E.,
Gertz
M.A.,
Kumar
S.K.,
Lacy
M.Q.,
Dingli
D.,
Buadi
F.K.,
Improved outcomes for newly diagnosed AL amyloidosis between 2000 and 2014: cracking the glass ceiling of early death. Blood.
2017;
129
(15)
:
2111-9
.
View Article PubMed Google Scholar -
Kumar
S.,
Dispenzieri
A.,
Lacy
M.Q.,
Hayman
S.R.,
Buadi
F.K.,
Colby
C.,
Revised prognostic staging system for light chain amyloidosis incorporating cardiac biomarkers and serum free light chain measurements. Journal of Clinical Oncology.
2012;
30
(9)
:
989-95
.
View Article PubMed Google Scholar -
Katzmann
J.A.,
Kyle
R.A.,
Benson
J.,
Larson
D.R.,
Snyder
M.R.,
Lust
J.A.,
Screening panels for detection of monoclonal gammopathies. Clinical Chemistry.
2009;
55
(8)
:
1517-22
.
View Article PubMed Google Scholar -
Rubinstein
S.M.,
Stockerl-Goldstein
K.,
How to Screen for Monoclonal Gammopathy in Patients With a Suspected Amyloidosis. JACC. CardioOncology.
2021;
3
(4)
:
590-3
.
View Article PubMed Google Scholar -
Knight
D.S.,
Zumbo
G.,
Barcella
W.,
Steeden
J.A.,
Muthurangu
V.,
Martinez-Naharro
A.,
Cardiac Structural and Functional Consequences of Amyloid Deposition by Cardiac Magnetic Resonance and Echocardiography and Their Prognostic Roles. JACC: Cardiovascular Imaging.
2019;
12
(5)
:
823-33
.
View Article PubMed Google Scholar -
Cheng
Z.,
Zhu
K.,
Tian
Z.,
Zhao
D.,
Cui
Q.,
Fang
Q.,
The findings of electrocardiography in patients with cardiac amyloidosis. Annals of Noninvasive Electrocardiology.
2013;
18
(2)
:
157-62
.
View Article PubMed Google Scholar -
Gertz
M.A.,
Li
C.Y.,
Shirahama
T.,
Kyle
R.A.,
Utility of subcutaneous fat aspiration for the diagnosis of systemic amyloidosis (immunoglobulin light chain). Archives of Internal Medicine.
1988;
148
(4)
:
929-33
.
View Article PubMed Google Scholar -
Merlini
G.,
Wechalekar
A.D.,
Palladini
G.,
Systemic light chain amyloidosis: an update for treating physicians. Blood.
2013;
121
(26)
:
5124-30
.
View Article PubMed Google Scholar -
Palladini
P.M. Giovanni,
Giampaolo Merlini. Management of AL amyloidosis in 2020. Blood.
2020;
136
(23)
:
2620-7
.
View Article Google Scholar -
Garcia-Pavia
P.,
Rapezzi
C.,
Adler
Y.,
Arad
M.,
Basso
C.,
Brucato
A.,
Diagnosis and treatment of cardiac amyloidosis. A position statement of the European Society of Cardiology Working Group on Myocardial and Pericardial Diseases. European Journal of Heart Failure.
2021;
23
(4)
:
512-26
.
View Article PubMed Google Scholar -
Muchtar
E.,
Dispenzieri
A.,
Gertz
M.A.,
Kumar
S.K.,
Buadi
F.K.,
Leung
N.,
Treatment of AL Amyloidosis: Mayo Stratification of Myeloma and Risk-Adapted Therapy (mSMART) Consensus Statement 2020 Update. Mayo Clinic Proceedings.
2021;
96
(6)
:
1546-77
.
View Article PubMed Google Scholar -
Lilleness
B.,
Ruberg
F.L.,
Mussinelli
R.,
Doros
G.,
Sanchorawala
V.,
Development and validation of a survival staging system incorporating BNP in patients with light chain amyloidosis. Blood.
2019;
133
(3)
:
215-23
.
View Article PubMed Google Scholar -
E. Muchtar,
S.K. Kumar,
M.A. Gertz,
M. Grogan,
O.F. AbouEzzeddine,
A.S. Jaffe,
A. Dispenzieri,
Staging systems use for risk stratification of systemic amyloidosis in the era of high-sensitivity troponin T assay. Blood.
2019;
133
(7)
:
763-6
.
View Article Google Scholar
Comments
Article Details
Volume & Issue : Vol 10 No 11 (2023)
Page No.: 6049-6056
Published on: 2023-11-30
Citations
Copyrights & License
This work is licensed under a Creative Commons Attribution 4.0 International License.
Search Panel
Pubmed
Google Scholar
Pubmed
Google Scholar
Pubmed
Google Scholar
Pubmed
Search for this article in:
Google Scholar
Researchgate
- HTML viewed - 3026 times
- PDF downloaded - 1078 times
- XML downloaded - 81 times
