

Copyrights: Hieu Ngo Trong, Hung Le Van Manh, Vy Thanh Dang, Nguyen Ho-Thao Nguyen, Phuc Van Pham, 2020. License: This work is licensed under a Creative Commons Attribution 4.0 International License.
Abstract
Immunotherapy, especially immune cell-based therapy, is a strategy for cancer treatment that has over the past decades focused on novel modifications and targets. In recent years, adoptive cell immunotherapy has continuously evolved, with studies of different platforms utilizing different immune effector cells to kill a variety of cancer cells. This review summarizes the various kinds of immune cells which have been used in adoptive cell therapy (ACT), including natural killer cells, cytokine-induced killer cells, T cells, and engineered immune cells. Most reports have shown that ACT can induce tumor regression, both in animal studies and clinical trials. However, the high cost of ACT is the greatest disadvantage of this strategy. Moreover, the efficacy of treatment is variable among patients. To reduce these disadvantages, off-the-shelf immune cells are considered the best solution to reduce the cost while maintaining the efficacy of treatment. In this review, we will discuss the potential of various kinds of immune cells for development as ``off-the-shelf'' immune cells for use in adoptive cell therapy, based on their unique advantages.
INTRODUCTION
Immunotherapy, especially cellular immunotherapy, has led to a renaissance in immunotherapy and has revolutionized cancer remedy options. Cancer immunotherapy is a strategy which uses the antitumor properties of the immune system to fight cancer. The concept of manipulating the immune system to combat cancer relies on the immune surveillance of malignant cells during cancer progression or abnormal cell growth. The field of cancer immunotherapy was initiated back in the 1890s in a study by W.B. Coley. Briefly, in his study, tumor regression was observed after injecting heat-killed streptococci at the tumor site of a patient with bone sarcoma1. To date, the immunotherapy field has achieved several milestones, including the approval of cytokine therapy (e.g. interleukin (IL)-2 and interferon (IFN)-alpha), immune checkpoint blockade (e.g. anti-cytotoxic T-lymphocyte-associated protein (CTLA4) monoclonal antibodies and anti-Programmed Death 1 (PD-1)/Programmed death-ligand 1 (PD-L1) antibodies), and cell-based immunotherapy (e.g. vaccine and adoptive cell therapy (ACT)).
Immunotherapy, especially ACT, has brought novel remedy options for cancer treatment. The development of cellular immunotherapy has included a long history of successfully discovering effective immune effector cells and appropriate cancer cell targets. Indeed, ACT is a promising cancer treatment approach that exploits the natural antitumor properties of human immunity to eliminate cancerous cells2. Lymphocytes are obtained from the patient (autologous cells) or from healthy donors (allogeneic cells), expanded ex vivo, and then passively transfused to patients3. This strategy can theoretically produce an adequate amount of effector immune cells and enhance the recipient's antitumor capacity by bypassing the tolerance of host immunity to tumor4. An early attempt of harnessing the immune function to eliminate cancer cells was demonstrated in 1953 by Mitchison in a mouse model; transplantation of an “educated” mouse lymph node to a naïve one showed therapeutic efficacy5. The first ACT clinical trial was reported in 1988 using tumor-infiltrating lymphocytes to treat patients with melanoma; later, in 2002, the development of pre-conditioning regimens significantly enhanced ACT6. More recently, there are been greater advances in cellular Immunotherapy, including the FDA-approved clinical applications of two chimeric antigen receptor (CAR)-T cell therapies (Tisagenlecleucel (KYMRIAH) and axicabtagene ciloleucel (YESCARTA) in 2019) for the treatment of relapsed or refractory large B cell lymphoma.
"Off-the-shelf" immune cells offer an ideal solution to universalize the application of cellular immunotherapy. This platform can solve the current problem of autologous immunotherapy, including cost, time and therapeutic efficiency. Note that the cost for a CAR-T infusion starts at $475,000 (the total cost with standard therapy can reach $968,800), and the cost for a therapy based on tumor-infiltrating lymphocytes (TILs) in the USA is from $97,600–$168,4407. Thus, only a small portion of patients can afford the cost of this state-of-the-art immunotherapy. Moreover, the major disadvantages of the above treatments are the lack of T cell production in cancer patients and the patients’ weakened immune system. The extent and obstinacy of antigenic stimulation are exposed during chronic infections and cancer, which leads to the compromise of T cell function; dysfunctional T cells fail to eradicate infection and cancer effectively8. Therefore, T cells derived from patients are often insufficiently generated to be utilized for cancer treatment.
The off-the-shelf strategy allows scale-up and cryopreservation to reduce production and process time, and to allow immediate availability and repeat transfusions. Furthermore, this strategy minimizes clinical trial data heterogeneity, creating an optimized standard for treatment. Altogether, the "off the shelf" immunotherapy has raised the hope of significantly improving cancer survival and revitalizing cancer immunotherapy. In this review, we cover the current strategies of adoptive cell immunotherapy (using natural killer cells, cytokine-induced killer cells, T cells, and engineered immune cells) and discuss the "off-the-shelf" approach for each type of cell.
| Immune cells | Description | Mechanisms | Advantages | Disadvantages |
| TILs | Natural T lymphocytes extracted from the tumor biopsy | Recognition of TAA through MHC-I complex by conditioned TCRs | TAA specificity and recognition | Restricted to MHC-I complex Low expression of antigenic peptides in specialized cells |
| Natural killer cells | Natural killer cells extracted from the peripheral blood or umbilical cord blood | Based on the expression of self-MHC class I, which suppresses NK cell activity through engagement with a set of killer cell immunoglobulin-like receptors (KIRs) | MHC-independent cytotoxicity | Non-specific killing |
| Cytokine induced killer cells | CIK cells can be easily expanded from mononuclear cells (MNCs) from peripheral blood, bone marrow, or umbilical cord blood | The cytotoxic function of CIK cells heavily depends on the expression of activating receptor NKG2D and NKG2D-associated adaptor molecule, disulfide adaptor protein 10 (DAP10) | MHC-independent cytotoxicity | Non-specific killing |
| TCR-T cells | T lymphocytes modified to express a tgTCR | Recignition of tumor antigenic peptides by tgTCR on MHC-I complex | Augmented specificity in targeting TAAs | Restricted to MHC-I complex and HLA-A2 patients tgTCR genetic mis-match with native TCR |
| CAR-T cells | Engineered T cells expressing an antibody-binding scFv exodomain fused with a CD3z chain intracellular domain via a transmembrane domain | CAR domain recognizes and binds to specific TAAs in a MHC-independent manner | Highly specific modified T cells | The cell prepararion is complex Cytokine release syndrome largely |
T LYMPHOCYTES
Lymphocytes are found in healthy tissues and blood, as well as found penetrated in tumor tissues. Accumulation of T lymphocytes in tumor tissues has been demonstrated to be correlated with a favorable prognosis, thus indicating their antitumor function and, in turn, emphasizes the interest in T lymphocyte strategies for cancer treatment8, 9, 10. Following the approval of IL-2 as a therapeutic agent, lymphokine-activated killer (LAK) cells were then introduced, as they were lymphocytes that could be grown in culture with IL-2. Despite being easily isolated and expanded ex vivo, LAK cells failed to achieve prolonged persistence in a patient with metastatic cancer, even with a high dose of IL-2 11.
Another ACT strategy is the use of TILs. A heterogeneous population of lymphocytes (including T cells and natural killer (NK) cells) was obtained from patient tumor tissue and expanded ex vivo12. These tumor-isolated lymphocytes contained tumor-specific T cell subsets and exhibited a stronger antitumor effect than peripheral blood-derived lymphocytes or LAK cells 13. The presence of TILs in primary melanoma has become a hallmark for prognosis; the high intra-tumoral level of CD8+ T cells over regulatory T cells (Tregs) showed greater informative value for prognosis and for success of immunotherapeutic regimens 14. Originally, TILs were isolated from metastatic melanoma and grown in vitro in IL-2 supplemented medium. Before TIL treatment, patients received non-myeloablative lymphoid-depleting chemotherapy; patients received subsequent IL-2 following TIL infusion to support in vivo survival of TILs and clinical efficacy15. TIL strategy is aimed at re-functionalizing impaired resident tumor-specific T cells, which are inactive in the tumor microenvironment.
TILs was first experimentally introduced in 198316. A study in 1988 demonstrated the safety and feasibility of TIL therapy; there were reports of toxicities associated with the administration of IL-2 but no toxicities were attributed to TIL infusion 17. Moreover, only melanoma-isolated TILs showed specific in vitro lytic activity against autologous tumors. A report on metastatic melanoma patients indicated that objective response (OR) was observed in 11/20 patients (55%); 2 of them responded to TILs after IL-2 treatment failure 18. Notably, no patients died upon treatment but reversible side effects resulted from IL-2 infusion. In 2011, a clinical trial was conducted to evaluate 93 patients with progressive metastatic melanoma following previous systemic treatment 19. The overall response rate (RR) was higher in the group treated with TILs and chemotherapy plus total body irradiation (TBI), compared to that without TBI (50% compared to 72%, respectively). In addition, 20/93 (22%) patients experienced complete response (CR), and 19/93 (20%) patients showed complete regression for more than three years. A recent systematic review and meta-analysis study showed that treatment with high-dose IL-2 plus TILs was comparable with checkpoint-blockade Immunotherapy (CBI)20. TILs offered a second-line therapy with acceptable responses for patients who failed to respond to CBI treatment and, more importantly, had fewer adverse events (AEs) and greater modification capabilities compared to CBI treatment alone. However, data suggest that tumors which escape from CBI might also escape from TILs using the same resistance mechanisms. CBI-naïve patients responded well to TIL treatment, with a median response duration of 69.1 months, compared to 4.8 months, in the case of patients with prior exposure to CBI21. Furthermore, a higher number of TIL infusion correlated with more favorable responses, but the effects were only recorded in CBI-naïve patients. Besides, a recent study demonstrated the feasibility of TILs in M1c melanoma patients; a 33% OR was achieved in patients with both treated and untreated brain metastasis 22. Data indicated that TILs that were safely localized to brain could mediate disease regression; however, therapeutic efficiency was limited and required additional treatment.
In addition to their success in melanoma cancer treatment, TILs have also been successful for other cancer types. Sanja Stevanovic et al. reported that 2 of 29 patients with HPV-associated epithelial cancers achieved CR following TIL treatment 23. Til the time of the publication, the 2 patients on the study survived out to 67and 53 months, respectively, following treatment. In a phase I clinical study with TILs following chemo-radiotherapy for advanced nasopharyngeal carcinoma (NPC), 19/20 (95%) of patients were reported with OR, and 18 patients experienced 12-month disease-free survival (DFS) or longer after treatment 24. In a 2018 report, in a case of chemo-refractory metastatic breast cancer, the patient achieved complete durable responses and survived for greater than 22 months after receiving TILs in combination with IL-2 and CBI 25. Therefore, those results demonstrate the promising potential of TILs for advanced cancer treatment.
Currently, 13 clinical trials with autologous TILs are supported by the National Cancer Institute (NCI). Although several studies have been focused on different types of solid tumors, the application of TILs is still restricted to melanoma, and its efficiency is limited by unsuccessful ex vivo expansion, scale-up, and storage. Moreover, TIL infusion requires a non-myeloablative lymphodepleting regimen and co-administration of IL-2, which are both associated with life-threatening adverse effects 12, 26. Data on TIL treatment is highly variable and the use of TILs is also limited due to their high cost. Overall, TILs still represent a promising approach as a personalized immunotherapy for future cancer treatment; further strategies for expansion and modification are under investigation to enhance the efficiency of treatment 15, 27.
NATURAL KILLER (NK) CELLS
Natural killer (NK) cells were first identified in 1975 as effector lymphocytes which belong to the innate lymphoid cell family 28, 29. Unlike T cells, NK cells can specifically recognize tumor cells by a unique mechanism that is different from the engagement of T cell receptor (TCR) on T cells with antigen bound to Major Histocompatibility Complex (MHC) molecules on target cells. In fact, NK cells do not require any activation to kill harmful cells that are missing MHC I markers (which would be unrecognizable by CD8+ T cells). Thus, the ability of NK cells to detect and efficiently kill abnormal cells, missed by other immune cells, lends support to their important role in immune surveillance. Their role in tumor suppression has also been demonstrated in mouse models and, importantly, in a prospective study with more than 3,500 participants over 11 years 30. The presence of infiltrating NK cells in tumor tissues has been correlated to improved prognosis 31, 32, 33. In blood, NK cells represent 10-15% of circulating lymphocytes. They are characterized by the absence of TCR/CD3 and the presence of neural cell adhesion molecules (NCAM/CD56). NK cells are divided into two main populations: immature CD3−CD56brightCD16− cells and mature CD3−CD56dimCD16+ 34, 35. Immature NK cells are responsible for cytokine production and immune regulatory function, while mature NK cells are responsible for cytotoxicity function.
The antitumor properties of NK cells are regulated by complex signaling, based on the balance of inhibition and activation signaling from different cell surface receptors 36, 37, 38. The principle of NK-mediated cytotoxic mechanism against infected/abnormal cells is based on the expression of self-MHC class I, which suppresses NK cell activity through engagement with a set of killer cell immunoglobulin-like receptors (KIRs). KIR-inhibitory receptor family is expressed on and is a marker of mature/functional NK cells. Mature NK cells are formed by a process called "licensing" or "education", which involves MHC and KIR interaction, contributing to fully functional NK cell development. In the presence of MHC class I molecules, KIR recognizes and transduces inhibitory signals, subsequently suppressing NK cell function. In contrast, in the absence of MHC class I as well as MHC-KIR engagement, known as "missing-self", the inhibitory signal by KIR is suppressed39. Self-reactive NK cells develop an anergic state, leading to incompetent function. Thus, NK cells mediate cytotoxicity toward cancer cells by "missing-self" mechanism, recognizing alteration of self-MHC on cancerous cells. Indeed, NK cells and CTLs represent a loophole in the immune system in that NK cells can replace the role of T cells when tumor cells alter MHC expression. The cytotoxic function of NK cells also requires the addition of stimulation signals. Stress-induced self-ligands, including MHC class I polypeptide-related sequence A/B (MICA/B) and UL16-binding proteins (ULBPs), are upregulated in stressed, infected or DNA-damaged cells, including cancerous cells. These ligands are detected by the activating NKG2D receptor, thereby shifting the balance of the NK cell activation state and inducing cytotoxic function. Other activating receptors involved in the cytolytic activity of NK cells include those of the natural cytotoxicity receptor family (e.g. NKp30, NKp46 and NKp44), and the DNAM-1 adhesion molecule4041.
Several clinical studies of autologous NK cell therapy have been conducted. Adoptive IL-2-activated autologous NK cell transfer has demonstrated safety in patients but also enhancement of NK cell activity in patients with advanced cancers 42, 43. Autologous NK cells can persist for up to months. However, the cells failed to suppress tumor progression and expressed low levels of NKG2D receptor 44, possibly due to KIR/KIR-L matching and "self" inhibition signals 45, 46. Other factors which contribute to the low antitumor effects include competition for cytokines and Treg development 47, 48. Several strategies have been proposed to improve therapeutic efficiency with autologous NK cell therapy, including ex vivo-primed NK cells with irradiated human feeder cell line, and inclusion of heat shock protein 70 (HSP70) in combination with immunoglobulin G-1 (IgG1) 49, 50. Autologous NK cells cultured with anti-CD16 showed improvement in 5-year progression-free survival (PFS) (51.1% vs. 35%, p = 0.044) and overall survival (OS) (72.5 vs. 51.6, p = 0.037) of stage IIIA-C colon cancer patients 51. A combination of HSP70-activated autologous NK cells and anti-PD-1 antibody improved survival of patients in stage IIIB non-small-cell lung carcinoma (NSCLC) patients; no cancer progression was detected out to 33 months 52.
Since the cytotoxic function of NK cells is mediated by an inhibitory signal from KIR/KIR-L engagement, the allogeneic approach presents KIR mismatch, thus suppressing inhibitory signals 53. Clinical studies have demonstrated antitumor activity based on KIR-mismatch of allogeneic NK cells. In an early study on haploidentical hematopoietic stem-cell transplantation (HSCT) for acute myeloid leukemia (AML) patients, alloreactive NK contributed to lowering the risk of relapse, thus avoiding graft rejection without graft-versus-host disease (GvHD) 54, 55, 56. Furthermore, patients who received allogeneic HSCT were reported to show NK lymphocyte reconstitution in peripheral blood. It is suggested that allogeneic NK cells prevent GvHD through cytolytic effects on T cells and dendritic cells (DCs), thus minimizing GvHD symptoms. The first clinical trial by Miller in 2005 demonstrated the feasibility and safety of allogeneic NK cell therapy for cancer patients; 5/19 patients with poor-prognosis AML showed complete hematologic remission without risk of severe adverse effects or GvHD 57. Haploidentical NK cells followed by IL-2 subcutaneous injection could persist up to one month in patients who received prior high-dose Cyclophosphamide and Fludarabine (Cy/Flu) conditioning regimen. Besides, a high level of endogenous IL-15 by Cy/Flu conditioning treatment favors in vivo expansion of NK cells and contributes to cancer remission in patients. Allogeneic NK cell infusion followed by stem cell transplantation and non-myeloablative condition regimens showed potential graft-versus-tumor (GvT) effect without toxicity58. Following the success of the first clinical trial, safety concerns and desire for enhanced clinical efficacy prompted further investigations in hematological malignancies59, 60, 61 and solid tumors, including NSCLC, ovarian, and breast cancer 62, 63, 64. Allogeneic NK cells showed antitumor effects against solid tumors in a study using allogeneic stem cell transplantation and low-intensity conditioning. Furthermore, the allogeneic NK cells exhibited the ability to infiltrate tumor tissues and cross the blood-brain barrier. In 2010, a study by Iliopoulou et al. showed that 2-4 doses of 2.9 x 107 NK cells/kg/dose were well-tolerated by the stage IIIB-IV NSCLC patients, without any symptoms of systemic side effects or GvHD 63. Moreover, among the 15 enrolled patients, 2 experienced partial response (PR) and 6 had stable disease (SD), with a median follow-up of 22 months. A study on malignant lymphoma patients showed that 8/17 patients developed stable disease, and 9/17 patients showed progressive disease (PD). Also, up-regulation of NKG2D on CD8+ T-cells and increase in chemokine production were reported 62. Interestingly, it was demonstrated that higher numbers of incompatible KIR led to better PFS in terms of clinical outcome.
Another source of allogeneic NKs for anti-cancer therapy is the umbilical cord blood (UCB). In UCB, NK cells account for 30% of whole blood, compared to 10% in peripheral blood, while bearing a similar ratio of CD56dim to CD56bright phenotype as that of peripheral blood. Moreover, the low level of T/NKT cells lowers the risk of GvHD. The use of UCB allows highly efficient NK isolation from the blood by a single step of positive selection for CD56. UCB-based HSCT showed safety in patients with hematologic malignancies, and donor NK cells were rapidly reconstituted upon transplantation. A report on adoptive transfer of UCB-derived NK cells showed that in 350 units of blood, researchers could select HLA-matched UCB partially for 10 AML patients in trials with both KIR receptor ligand mismatch and KIR B haplotype 65.
The use of UCB-NK cells lowers the risk of contamination with circulating tumor cells from patients and viral transmission from a donor. It has been reported that the low production of granzyme B, perforin, IFN-gamma, and tumor necrosis factor (TNF)-alpha limit the cytotoxic function of UCB-NK cells. Upon stimulation by cytokines, such as IL-2, IL-15 and IL-18, UCB-NK cells acquired cytotoxic function66. A report on the CD56bright population of UCB-NK cells showed that upon stimulation by IL-2 and IL-18, there was a significant enhancement of IFN-gamma production 67. NK cells expanded with K562-based artificial antigen-presenting cells (aAPCs) under GMP conditions not only showed upregulated expression of perforin and granzyme B, but also displayed strong cytolytic ability against multiple myeloma, B-cell non-Hodgkin lymphoma, and leukemia cancer models 68, 69, 70, 71. Using a gas-permeable culture system following GMP compliance, NK cells were expanded up to 2,389-fold from frozen UCB and 1848-fold from fresh UCB, yielding > 95% of the cell population. In a study in 2017, a phase I clinical trial recruited 12 patients with advanced myeloma and divide the subjects into four cohorts for treatment with four different dosages 72. UCB was chosen based on 4/6 HLA matched loci, and NK cells were expanded from the cryopreserved UCB under GMP conditions in a gas-permeable bioreactor. The UCB-derived NK cells were expanded with IL-21-expressing aAPCs in IL-2, and after CD3-depletion, accounted for 98.9% of the cell population prior to infusion. Patients received lenalidomide (10 mg) and melphalan (200 mg/m2) prior to UCB-NK treatment. Results showed that doses of UCB-NK cells up to 1 x 108 cells/kg/dose were well-tolerated, and there were no recorded toxicities or GvHD. Moreover, 10/12 patients experienced very good PR, with a median follow-up of 21 months. Interestingly, UCB-NK cells acquired the expression of CD16, KIR, NKG2D, NKp30 and inhibitory receptor NKG2A following in vivo infusion, thereby suggesting that pre-treatment with the combination therapy (of lenalidomide and melphalan) enhances the clinical efficiency of NK cells.
On the other hand, UCB is a rich source of CD34+ hematopoietic stem cells (HSCs) and hematopoietic progenitor cells (HPCs). Cryopreserved UCB showed no alteration in phenotype and cytotoxic function while CD34+-derived NK cells retained their function compared to fresh UCB; moreover, upon stimulation by cytokines, the latter cells expressed matured cell markers CD16 and KIR, and exhibited strong efficiency at killing cancer cell both in vitro and in vivo 73, 74, 75. Dolstra et al. succeeded in expanding NK cells up to 2000-fold from UCB-CD34+ cells in clinical grade conditions, reaching > 90% purity 76. UCB-derived CD34+-NK cells first demonstrated promise as a cellular immunotherapy in 10 old-aged patients with a poor prognosis of AML 65. In terms of safety, infusion of UCB-derived NK cells, with cyclophosphamide and fludarabine pre-conditioning, was well-tolerated by the patients; there were no observed immediate toxicities, GvHD, or cytokine release syndrome. Indeed, 4/10 patients experienced DFS of 60, 52, 22 and 16 months, while the other 6 patients had a median survival of 12 months. Some patients also received non-myeloablative conditioning of Cy/Flu, and without co-administration of exogenous IL-2. Interestingly, this treatment resulted in higher endogenous IL-15 concentration in the blood, thus favoring the maturation of UCB-derived NK cells post-infusion, and upregulating CD16 as well as KIR. Moreover, there was no significant Treg proliferation in patients following non-IL-2 treatment. The latter results correlate with other studies, particularly with in vivo studies in which co-injection of IL-15, or in combination with monoclonal antibodies, showed enhancement of patient survival 73, 74. A study of metastatic colorectal cancer (mCRC)-bearing mice showed that UCB-derived NK cells induced a significantly higher cytotoxic effect than peripheral blood-derived NK cells 73. A combination of IL-15 and UCB-derived NK cells led to upregulation of CD16 and KIRs in NSG-leukemia mouse model 74, 75. Data indicated that conditioning regimen or co-administration of IL-15 favors maturation of UCB-derived NK cells in in vivo conditions, thereby enhancing NK cell cytotoxicity.
Along with UCB-NK and peripheral blood-NK cells, the NK cell line is an alternative approach that has also been extensively studied due to availability and accessibility. Different NK cell lines have been isolated from leukemia and lymphoma patients, and grown in vitro 77. NK-92 is the most popular NK cell line from non-Hodgkin’s lymphoma patients. It has been extensively studied in preclinical and clinical trials, and approved for clinical trials by the US FDA as a cell immunotherapy source. NK-92 cells were expanded in vitro in IL-2 dependent conditions, and scaled up in ex vivo conditions following cGMP compliance 78, 79. These cells lacked KIR expression and highly expressed NKG2D, resulting in KIR mismatch and superior antitumor effect 80. Preclinical studies showed that irradiated NK-92 is safe, without risk of immunogenic or tumorigenic potential and GvHD. The NK-92 cell line also possesses an antitumor effect against a broad spectrum of human cancers in xenograft models 80, 81, 82, 83. However, these cells lacked CD16, and thus unable to mediate cytotoxicity by the ADCC pathway, limiting their cytotoxicity.
Several phase-I trials have demonstrated the feasibility of the NK-92 cell line for cancer treatment. Several studies reported that infusions of irradiated NK-92 were well-tolerated, with no long-term toxicities recorded, and the incidence of AEs was rare 82, 84, 85, 86. The studies have included advanced cancer patients, with doses up to 1 x 1010 cells/m2 that were well-tolerated without severe AEs. General reactions were reported as mild and transient. Overall, severe toxicities of grade 3/4 were reported. However, their frequency was low. Clinical responses were reported in some cases, even though patients were under advanced disease status. Briefly, 75% of lung cancer patients (3 of 4 patients) achieved positive clinical response and SD, and NK-92 cells persisted in vivo in blood up to 48 h after infusion in 2 patients 87. A recent clinical study on refractory and relapsed AML showed that although no significant clinical responses were reported, adoptive transfer of NK-92 demonstrated safety and potential 86. In this study, 3 of 7 patients were reported with transient activity while no dose-limiting toxicities or grade 3-4 toxicities were observed. Furthermore, there was no changes in characteristics/properties of the recipient’s lymphocytes following NK-92 cell infusion. According to Clinicaltrials.gov, 14 clinical trials with NK-92 are underway, in recruitment phase, enrollment by invitation, or active (though not recruiting) status. There were also three completed clinical trials with patients bearing hematological malignancies and solid tumors. Compared to autologous NK cells, NK-92 cells have the properties for "off-the-shelf" production. NK-92 represents a readily available source of cell and meets the standard for a cGMP-compliant cell bank. Indeed, primary NK cells are highly variable and inefficient to be scaled up, while the NK-92 cell line can be scaled up and cryopreserved for repeated transfusions.
Moreover, NK-92 cells are easily modified by genetic engineering. Extensive studies have tried to improve the antitumor activity of NK cells and overcome the limitation of lack of CD16 expression 88. High-affinity NK cells (haNK), which are engineered NK-92 cells with CD16A receptor and endogenous IL-2, have been authorized for several clinical trials against solid tumors35, 53, 80. Regarding the use of target-activated NK or CAR-NK cells, engineered NK-92 cells have been investigated for glioblastoma, neuroblastoma, NSCLC, myeloma, prostate cancer, and breast cancer in preclinical and clinical trials. Also, anti-CD19-NK-92 has been investigated for hematological malignancies and anti-CD33 has been evaluated for AML patients. However, NK-92 cells are derived from lymphoma cancer cells, requiring irradiation before transfusion to prevent their proliferation in recipients, thus limiting their cytotoxic function and long-term efficiency.
Both autologous and allogeneic NK cell therapies bear different advantages and disadvantages. Both still have potential in immunotherapy applications. Autologous NK cells show safety without severe AEs. However, their low efficacy and the possibility of contamination from circulating tumor cells (CTCs) represent two major problems that need to be considered. On the other hand, allogeneic NK treatment results in a more favorable outcome. Similarly, allogeneic NK cells have problems, namely that they are linked to more AEs and potential of GvHD from T cell contamination. Conditioning regimens and administration of exogenous cytokines are required to prevent rejection and long-term effects of allogeneic NK cells. The lack of available data and controversial results also limit the application of allogeneic NK cells. However, the allogeneic approach provides an available source of cells and the potential of an "off-the-shelf" solution for immunotherapy. Overall, both auto- and allo-NK approaches hold promise as future platforms of immunotherapy. However, more developments are needed to enhance their therapeutic use.
CYTOKINE-INDUCED KILLER (CIK) CELLS
Cytokine-induced killer (CIK) cells are a heterogeneous population derived from CD8+ T cells, and are best known as CD3+CD56+ natural killer T (NKT) cell phenotype. CIK cells can be easily expanded from mononuclear cells (MNCs) from peripheral blood, bone marrow, or umbilical cord blood. CIK cells were reported to show a high expansion rate and strong cytotoxicity compared to LAK cells 89. CIK cells are expanded ex vivo following the standard protocol using anti-CD3 antibody, IFN-gamma, and IL-2 90. Over 14-21 days, CIK cells can expand up to 1000-fold, and consist of two main subsets (CD3+CD56- and CD3+CD56+), and a low level of CD3−CD56+ NK cells. The antitumor activity of CIK cells is independent of MHC molecule and is attributed to the CD3+CD56+ population 89, 91. The cytotoxic function of CIK cells heavily depends on the expression of activating receptor NKG2D and NKG2D-associated adaptor molecule, disulfide adaptor protein 10 (DAP10). NKG2D is upregulated in the presence of IL-2 in in vitro culture. Recently, the replacement of IL-2 by IL-15 resulted in upregulated NKG2D expression and enhanced cytotoxic function of CIK cells 92, 93, 94, 95. Lymphocyte function-associated antigen 1 (LFA-1) and intracellular cell adhesion molecule 1 (ICAM-1) also play important roles in mediating the cytotoxic function of CIK cells through cell-cell contact89, 96. The cytotoxicity of CIK cells is controlled via FasL, perforin/granzyme B, as well as secretion of TNF-alpha and IFN-gamma 97, 98. Compared to NK cells, CIK cells lack inhibitory receptors (KIR, NKG2A/CD94) and show low expression of NK-specific activating genes (NKp30, NKp44, and NKp46) 99. NKp30 at low density in CIK cells has been demonstrated to play a role in the recognition and killing of lymphoma cells in response to IL-15-supplemented conditions 100, 101. Since CIK cells retain expression of TCR, engagement of TCR-MHC molecules augments the cytotoxic function of CIK cells; in contrast, interference in TCR engagement shows no impact 100. Several studies have focused on improving proliferation as well as cytotoxicity of CIK cells90, 102.
In vitro studies have shown that CIK cells demonstrate greater cytolytic effects compared to LAK cells against different cancer cell lines, including glioma, hepatocellular carcinoma, ovarian cancer, cervical cancer, lung cancer, lymphoma, primary AML, and chronic myelogenous leukemia (CML) 90, 103. Recent studies further indicated the effectiveness of CIK cells toward cancer stem cells and chemo-surviving cancer stem cells 104, 105, 106. In mouse models, CIK cell infusion promoted a prolonged survival of tumor-bearing mice. CIK cells induced minimal toxicity against hematopoietic precursors 107. A study on lymphoma-bearing mice showed that CIK cells could be detected in the liver and spleen within 16h after infusion. CIK cells could be detected at 72h after infusion at the tumor site and persisted up to 9 days later 108. Mice receiving CIK cell infusion showed tumor regression; with respect to survival, the mice showed no relapse up to 6 months after infusion. In contrast, untreated mice experienced severe PD. In 1999, the first phase I clinical trial of autologous CIK infusion was conducted in 10 patients, and demonstrated the feasibility and safety of autologous CIK-based cell immunotherapy. In that study, the only AE observed was grade 2 fever, which was ameliorated by the next day 109. In 2005, Thomas Leemhuis et al. conducted a phase I study consisting of 9 relapsed lymphoma patients. No immediate adverse reactions were recorded. In terms of treatment outcome, 2 patients showed PR and 2 achieved SD 110. In recent years, phase III clinical trials of autologous CIK cells have been conducted, with patients receiving autologous CIK immune-cell therapy showing improved PFS and OS. Moreover, severe AEs were similar for both control and treated groups, suggesting that there were no CIK-related severe adverse effects 111, 112, 113. Several autologous CIK studies have been conducted in solid and hematologic malignancies, indicating the therapeutic efficacy and safety of autologous CIK cells in high-risk patients90, 99, 102, 103, 114, 115, 116, 117.
Since CIK cell function does not depend on MHC recognition, approaches using allogeneic CIK cells have been extensively studied and reported in literature. Preclinical models have demonstrated low GvHD symptoms and potential cytolytic activity of CIK cells in an allogeneic setting 118, 119, 120. In in vivo mouse studies, infusion of 20 x 106 CIK cells was well-tolerated, while infusion of 2.5x106 splenocytes led to acute GvHD 121, 122, 123. Interestingly, infused CIKs produced high levels of IFN-gamma, which prevented the GvHD effect 122, 124. According to Sangiolo et al., the CD3+CD56− population in the allogeneic CIK cells were responsible for the GvHD effect; on the other hand, CD3+CD56+ cells maintained their cytotoxic function without causing GvHD 118, 120. Also, CIK cells expressed lower GvHD-homed molecules and had lower in vivo division rates, making them highly susceptible to apoptosis than naïve splenocytes. CIK cells also induced a lower risk of GvHD in the allogeneic approach.
Moreover, clinical studies have demonstrated the feasibility, safety, and efficacy of allogeneic CIK cells. The first phase I clinical trial used allogeneic CIK cells to treat 11 patients with relapsed hematological malignancies after HSCT. CIK cell dose was up to 12.4 x 106 cells/kg 125. Treatments were well-tolerated, and no immediate adverse effects were recorded during infusion. Among the 11 patients, 3 showed CR and 1 had SD. Acute GvHD was recorded in 4 patients; 2 of them developed chronic GvHD later. Interestingly, patients who developed GvHD had the most significant clinical responses. Following the success of the first phase I clinical trial, several phase I/II clinical studies have since been carried out to further investigate the clinical efficacy of allogeneic CIK cells in hematological malignancies 124, 126, 127.
Recently, a phase IIA clinical trial in 2017 evaluated allogeneic CIK cell therapy in 73 participants who experienced relapse after allogeneic HSCT. The treatment scheme included donor lymphocyte infusion (DLI), followed by allogeneic CIK infusion126. Among the 73 participants, 3 died before treatment due to disease progression and 60 patients received CIK infusion, with 43 of the 60 finishing the treatment course. Safety data indicated that allogeneic CIK administration was responsible for 4/12 cases of acute GvHD and 11 cases of chronic GvHD (10 of which occurred during or after finishing CIK treatment). CR was observed in 19/71 (26%) patients, PR observed in 3 (4%) patients, and SD in 8 (11%) patients. The proportion of patients with PFS for 1 and 3 years was 31% and 29%, respectively, whereas OS was 51% and 40%, respectively. A study in 2019 evaluated the efficacy of CIK infusion compared to DLI. Allogeneic CIK cell infusion resulted in a significantly lower risk of GvHD than that from DLI, and CR was achieved in 68% of patients treated with CIK therapy. Cumulative incidence of relapse (CIR) in CIK-treated patients was significantly lower than that of those receiving DLI, even though patients with CIK treatment had higher risk score and worse prognosis128.
In study of 303 recruited patients with six different solid tumors, 54 were diagnosed with NSCLC and chosen for further evaluation 129. It was reported that there was no significantly different response between autologous and haploidentical allogeneic treatment. The significant AEs included insomnia, fever, nausea, vomiting, and mild abdominal pain. In the 54 patients with stage IIIB-IV NSCLC, treatment with haploidentical allogeneic CIK cells resulted in better survival with a duration up to 48 months. The median survival time was 11 months (95% CI, 9.2‑11.8 months) compared to 8.0 months (95% CI, 9.2‑11.8 months) in the patient group with optimal support.
UCB has also been harnessed as a source of allogeneic CIK cells for clinical application. In preclinical studies, UCB-derived CIK cells showed a higher proliferation rate, with up to 500 x 106 cells per cord blood unit. The cells were also less susceptible to apoptosis in response to cisplatin treatment, and showed lower HLA expression, lower immunogenicity, and lower incidence of GvHD 130. Another study showed that UCB-CIK cells expressed a high level of cytotoxic markers, such as perforin, granzyme B, NKG2D, and chemokine receptor CCR6/CCR7, as well as elevated secretion of IL-2 and IFN-gamma 130, 131, 132, 133. A clinical study of 5 patients with relapsed hematologic malignancies after CBT demonstrated the feasibility of the approach 134. Patients received CB-CIK cell treatment with a median of 1.5 x 106 cells/kg. No acute adverse or delayed effects were observed, except for 1 patient with critical grade III GvHD. In another study (in 2011), 40 Chinese patients with advanced solid malignancies who failed the first-line chemotherapy treatment were recruited 133. The treatment regimen was second-line chemotherapy plus UCB-CIK and dexamethasone. Results showed that CIK cells plus chemotherapy significantly improved the clinical outcome of the patients. Moreover, a longer median survival time and time to progression were achieved in the combined treatment, without any significant CIK cell-related AEs. It is worthy to note that patients who developed GvHD achieved PR, which was correlated to previous data on allogeneic CIK cells. Thus, these data demonstrate the capability and potential of UCB-CIK cells in future clinical applications 125.
According to a report of the International Registry on CIK cells (IRCC) in 2015, 1,520 of 2,729 total patients were enrolled in 45 studies of CIK cell treatment; CIK cells were infused up to a maximal dose of 7.7 x 109 cells per infusion, with no severe adverse effects observed 115. Fever (from 37.5oC to 40oC) was the most common adverse effect which occurred in 40.09% of the treated patients; the other adverse effects included fatigue, headache, fever-related chills, rash, nausea, and vomiting. In terms of PFS and OS, 19 studies with 1,135 CIK-treated patients were accessed; 15 of those studies reported that CIK treatment resulted in significantly prolonged PFS and OS compared to the control group. Furthermore, 4 out of 5 studies reported that the quality of life (QOL) was significantly higher, as shown by the Karnofsky score after CIK treatment. The evaluation criteria, including fatigue, mental health and appetite, were improved in treated patients. This report also showed that across 4 studies with a total of 52 patients treated with allogeneic CIK transfusion, only 14 patients experienced GvHD. However, it is important to note that these patients responded well to corticosteroids. The database of clinical trials (http://www.clinicaltrials.gov) indicate that China has emerged as a country with extensive CIK cell therapy clinical studies. Moreover, the Chinese researchers have developed a literature database, i.e. VIP database of Chinese scientific and technological journals (http://oldweb.cqvip.com), of which numerous clinical studies on CIK cells have been included. A review in 2012 was conducted to summarize the Chinese publications on CIK-based clinical applications across 24 studies with 936 patients 116. The available information from 14 studies revealed the potential of CIK cells in immunotherapy. Indeed, CIK cell treatment achieved a total RR of 51.7% (291/563), with CR reported in 40 patients and SD reported in 135 patients. Data on overall survival rate (OSR) was revealed in 10 different publications. Based on the data, the 1-year OSR in CIK cell-treated patients was 72.5% (74/102), 2-year OSR was 66.3% (136/205), 3-year OSR was 75.5% (40/53), and 5-year OSR was 38.2% (42/110). According to the review, AEs commonly appeared as fever; patients responded well to anti-inflammatory drugs since fever was relieved after 24 h. Overall, patients showed improvement in QOL with improved sleep, body weight, and appetite, as well as less pain and fatigue.
In conclusion, we highlight the success of phase I and II clinical trials using autologous and allogeneic CIK cells derived from both peripheral blood and umbilical cord blood94, 114, 126, 127, 131, 135, 136, 137, 138, 139. Furthermore, CIK cells can be scaled up under GMP-compliant standards. Regarding clinical efficacy, CIK cells show promising results in the allogeneic setting in terms of cancer remission and safety. Several studies have tried to optimize and standardize the treatment protocol. However, there are limitations pertaining to the heterogeneous clinical studies, evaluation criteria of CIK cell treatment, cell source, and matching system. Thus, CIK cells are feasible as an "off-the-shelf" immunotherapy platform in the future.
TCR-T CELLS
T cells play a pivotal role in the human adaptive immune system 140. Besides their use as TILs, as mentioned earlier, T cells can be genetically modified ex vivo then infused back into patients for optimal anticancer effects. Engineered T cells, including TCR-engineered T (TCR-T) cells and CAR-T cells, have recently presented as one of the most promising immunotherapies against cancer 141.
The discovery of TCR in 1984 opened a new era for several immune-related research fields, including cancer immunotherapy142, 143. TCR-T cell therapy involves utilizing TCR with heterodimers composed of alpha and beta chains to recognize antigenic peptides presented by APCs via MHC molecules 144. Naturally-occurring TCRs have a low affinity for tumor antigens, leading to the inefficiency in tumor recognition and killing 145. Artificially designed TCRs can recognize a specific target of interest with higher affinity, resulting in a more favorable treatment outcome. Targeted antigen recognition then provides an activation signal via CD3 chains for T lymphocytes to perform their antitumor function 146.
One significant advantage of TCR-T cells is that they can recognize intracellular, surface, and neo-antigens, so long as those antigens are capable of being presented by an MHC molecule. Moreover, TCR-T cells are more promising than CAR-T cells in terms of solid tumor treatment due to better intratumoral penetration potential 147. In a systematic review by Zhang and Wang, of 84 clinical trials of TCR-T cells, it was demonstrated that the most targeted antigen was New York esophageal squamous cell carcinoma (NY-ESO-1), with melanoma being the cancer type most studied (33 clinical trials). Among those trials, only 16% were for the study of hematologic malignancies, as the therapeutic effect of TCR-T cells is predominantly strong in solid tumors 148.
On the other hand, there are disadvantages of using TCR-T cells. These include the large potential antigen pool, selection of specific tumor targets, and screening for optimal TCR recognition and affinity, all of which are labor-intensive and costly processes 147. Additionally, due to the required interaction between TCR and MHC for T cell activation, HLA alleles of the T cells and that of the patients must be matched in order to induce inhibition of cancer cells. If mismatch occurs, recognition of autoantigens can lead to GvHD 149. MHC-restricted fratricide of lymphocytes and TCR-mediated hematopoietic stem cell killing limit the clinical use of TCR-T cells 150. Autologous T cells are a great source for TCR-T cell immunotherapy due to the MHC-restricted antigen recognition 151. However, this cell source has several limitations, such as the compromised immune system of patients treated with chemotherapy, high production cost, and possible manufacturing failures. Also, patients with poor prognosis or relapsed tumor have limited wait time for being treated.
Off-the-shelf TCR-T cells are currently unavailable on the market. However, scientists are still investigating methods for expanding TCR-T cells for clinical use in a broader spectrum of patients with more reasonable prices and with more optimal outcomes. Given that HLA-dependent recognition provides TCR-T cells with a more extensive range of targets, it also presents a primary disadvantage of TCR-T cell therapy due to the limited patients with suitable HLA alleles. HLA-A*0201 has been the most frequently targeted HLA since it is most common in the Caucasian population. The use of a total of 4 therapeutic HLA class I alleles, including A*0201, A*0301, A*2402 and B*0702, can cover up to more than 90% of the Caucasian population in the US 152. In 2009, it was reported that the use of low-copy number retroviral vector- encoding both siRNA-targeting endogenous TCR and also siRNA-resistant TCR targeting human antigens MAGE-A4 or WT1- could result in elevated expression of tumor-specific antigen and enhanced cytotoxicity against tumor cells 153. Provasi et al. showed the first successful gene deletion in primary T cells by ZNFs to remove endogenous TCR and express engineered high-affinity TCR 154. The disruption of endogenous TCR was shown to prevent GvHD in treated patients.
Moreover, allogeneic T cells targeting minor histocompatibility antigen HA-1 have an advantage in leukemia due to their restricted high expression level in hematopoietic cells compared to nonhematopoietic cells. Therefore, those T cells can prevent both GvHD and off-target effects 155. This method involves the transplantation of allogeneic stem cells from an HA-1 negative donor into leukemic patients with HA-1 positivity, and HLA-A*0201 can be used for treatment in 15 to 20% of patients 156. Interestingly, Amelie Montel-Hagen et al. showed that pluripotent stem cells transduced with HLA-A*0201-restricted TCR specific for NY-ESO-1 peptides could develop into CD8+ T cells with antigen-specific cytotoxicity after being cultured in a 3-D artificial thymic organoid system. These cells also lacked endogenous TCR expression due to allelic exclusion, which gives the potential for “off-the-shelf” production of TCR-T cells 157.
Additionally, it was reported by Sarah Bonte et al. that hematopoietic stem and progenitor cells (HSPCs) without endogenous TCR expression could be transduced with antigen-specific TCR to develop into mature T cells after being cultured in an OP9-DL1 co-culture system. These cells expressed only one HLA-restricted antigen-specific TCR without the endogenous TCR; therefore, this method is also suitable for “off-the-shelf” TCR-T cell production. Moreover, the authors also noticed that HSPCs from UCB had faster maturation and higher cell yield compared to cells from peripheral blood of healthy donors and cancer patients 158.
CAR-T CELLS
CAR-T cells have recently been regarded as one of the most prominent anticancer therapies. Four generations of CAR-T cells have been established in the history of CAR-T cell evolution. The first generation of CAR-T cells is comprised of an extracellular binding domain, a hinge, a transmembrane domain, and an intracellular signaling domain (which is usually the CD3ζ chain of TCR) 159, 160. A single-chain variable fragment (scFv) derived from tumor antigen‐reactive antibodies is incorporated into the extracellular binding domain. This fragment can form specific binding to the over-expressed tumor antigen, which presents the most important characteristic of cancer cell-specific recognition. CD3ζ is another component that is present in all CAR generations. In the second generation of CAR-T cell, a co-stimulatory domain (such as CD28 or 4-1BB) is added to the already present CD3ζ chain in the first CAR-T generation 161. This additional domain helps CAR-T cells to improve proliferation, cytokine secretion, and in vivo persistence. Kymriah (Novartis) and Yescarta (Kite Pharma), produced based on this generation of CAR-T cell development, are 2 FDA-approved cell therapies for the treatment of B-cell acute lymphoblastic leukemia and relapsed large B-cell lymphoma 162. The third generation of CAR-T cells utilizes multiple costimulatory domains, e.g., CD28-41BB or CD28-OX40. These combinations result in superior expansion and longer persistence in treated patients 163, 164. The fourth generation of CAR-T cells, or TRUCK T cells, have an added transgenic cytokine and transgenic costimulatory ligand. The addition of cytokine can lead to more enhanced CAR-T activation, and can also induce the activation of innate immune cells to attack antigen-negative cancer cells 165, 166.
CAR-T cell therapy has been proven to be outstandingly effective against hematological cancers in clinical trials. Although autologous CAR-T cells are a great platform for immunotherapy due to longer in vivo persistence and absence of allogeneic reactions, some major drawbacks include high cost for personalized treatment, long manufacturing time with possible failures, and T cell dysfunction in cancer patients. Nonetheless, the application of "off-the-shelf" CAR-T cells in the future for cancer treatment holds great promise.
CAR-T cells can be derived from other allogeneic sources, including peripheral blood of healthy donors, UCB, induced pluripotent stem cells (iPSCs), or embryonic stem cells (ESCs). These allogeneic cell sources provide several benefits compared to the autologous cells from cancer patients. Firstly, these cell sources are readily available, which can speed up the process of cell manufacturing. Firstly, patients with advanced cancer stage who normally have limited time available for treatment can benefit from a short period of waiting time. Secondly, doctors and hospital technicians can establish a cell bank with different HLA subsets to treat the appropriate groups of patients to minimize GvHD. Thirdly, healthy immune cells harvested from healthy donors can be superior in cell function because they were not affected by chemotherapeutic agent treatment or dysregulated by tumor cells. Moreover, experts can select the donor with a suitable immune cell profile and health condition for optimal treatment outcome, based on quality control of the donor blood.
Transplantation of CAR-T cells derived from UCB is associated with lower chronic and acute GvHD, especially when an HLA-matched donor is not available. Thus, this treatment is urgently needed 167. Moreover, Julian Kwoczek et al. reported that T cells generated from UCB express a naive phenotype and show improved homing ability, which possibly contributes to the decrease in alloreactivity of transplantation 168. Besides, it has been reported that UCB-derived T cells express decreased nuclear factor of activated T cells-1 (NFAT1) expression, leading to reduced incidence of GvHD 169.
Additionally, iPSCs can be utilized for CAR-T cell generation 170. Allo-rejection of CAR-T transplantation can be minimized by using a bank of iPSC line with common HLA haplotypes. Moreover, gene editing methods, such as CRISPR-Cas9, can be used to remove the TCR gene to avoid GvHD. The established product is homogenous since it is derived from one master iPSC line, potentially improving the safety and efficacy of treatment.
GvHD is one of the most potential risks that must be prevented in allogeneic CAR-T cell transplantation. It has been reported that alloreactive αβ T cells are responsible for both acute and chronic GvHD 171, 172. Several strategies have been developed to minimize the risk of GvHD to enable the use of allogeneic CAR-T cells in "off-the-shelf" immunotherapy approaches. In patients who have relapsed cancer after stem cell transplantation, using CAR-T cells derived from the original donors presents a method to eliminate the risk of GvHD173. Another approach is utilizing virus-specific memory T cells to treat Epstein-Barr virus-associated cancers. This treatment type can reduce GvHD potential and is now under investigation in both preclinical and clinical settings 174. Additionally, the concept of integrating CAR into another cell type instead of αβ T cells has been widely studied by several research groups. NK cell, a cell type with well-known anticancer activity, is inactivated in several cases of cancer patients. Engineering a CAR onto NK cells can strengthen their cytotoxicity against cancer cells 175. As with NK cell therapy, the NK92 cell line can also be used as a target for CAR transfection under GMP conditions 87. Also, cord blood-derived NK cells have been engineered to express IL-15 and CD19-targeted CAR, leading to prolonged survival in the human body and enhanced anticancer activity 176. Another cell type that can be used is T lymphocytes expressing NK cell markers, i.e. NKT cells. A small subset of this cell type is called invariant NKT (iNKT) cells, which have the ability to regulate various immune responses, including acute GvHD 177. Indeed, iNKT cells can be modified to express CD19-targeted CAR and have been shown to exert cytotoxicity against lymphoma cells in preclinical models 178. One potential cell type to be engineered with CAR is the γδ T cell. As previously mentioned cells, γδ T cells are also capable of killing tumor cells, and they differ from αβ T cells in terms of their MHC-unrestricted anticancer mechanism 179.
Furthermore, macrophages, a composition of the innate immune system, have been proven to be able to carry antibody-directed CAR to kill cancer cells without causing GvHD 180. In order to prevent GvHD disease, another method that is under investigation is eliminating TCR in αβ T cells.
As previously mentioned, TCR in αβ T cells is the essential factor modulating GvHD response after transplantation. Although John S. Bridgeman et al. reported that TCR is required for the optimal response of CAR-T cells, Hiroki Torikai et al. have demonstrated the opposite 181. Briefly, Torikai et al. described that using Sleeping Beauty and zinc finger nucleases gene editing system, α and β TCR chains can be deleted to prevent GvHD. Together with the addition of CD19-targeted CAR, these CAR-T cells retained their specific cytotoxicity against CD19+ cancer cells. The genome editing tools, including TALEN and CRISPR, allow the knockout of endogenous TCR, which can prevent GvHD, and of CD52, which is important for tolerance against lymphodepleting agent Alemtuzumab after allogeneic stem cell transplantation 180. In an article published in Nature in 2017, Justin Eyquem et al. reported a strategy, incorporating CAR construct into the TRAC locus by CRISPR-Cas9, to introduce CAR and to inactivate TCR at the same time. This approach also minimizes the mutagenesis caused by false insertion and can be a great candidate to develop "off-the-shelf" CAR-T cell immunotherapy182. The first clinical trial result using TALEN-edited CAR-T cells in B-cell acute lymphoblastic leukemia (ALL) was reported by Waseem Qasim et al.183. In this study, 2 patients with relapsed refractory CD19+ B-cell ALL received lymphodepleting chemotherapy, anti-CD52 serotherapy, and a single-dose of allogeneic CD19 CAR-T cells. Both patients achieved molecular remission without significant GvHD. In another preliminary report of allogeneic CD19 CAR-T cell clinical trial, 5 out of 6 pediatric patients and 8 out of 10 adult patients achieved complete remission. Importantly, no GvHD was observed in any of the patients 184.
CONCLUSION
"Off-the-shelf" cell immunotherapy has opened a new era for the treatment of cancer patients. Since the beginning of immune cell therapy, a personalized approach has been well-practiced in clinics. However, due to the enormous amount of time and resources needed for preparation, this effective anticancer strategy has not reached many cancer patients. "Off-the-shelf" cell therapy outweighs personalized therapy in terms of availability, accessibility, cost-effectiveness, and technical simplicity. Several aspects of "off-the-shelf" immunotherapy still present some challenges, including ex vivo cell expansion, survival and proliferation of cells after Infusion, GvHD-inducing potential, and consistent manufacturing processes. With all the advantages mentioned above for “off-the-shelf" cell immunotherapy, there is great potential and need for scientists and pharmaceutical companies to work in cooperation to advance this revolutionized therapy and make it available to cancer patients worldwide in the future.
Abbreviations
ACT: Adoptive cell therapy
AE: Adverse event
ALL: Acute lymphoblastic leukemia
AML: Acute myeloid leukemia
APC: Antigen presenting cell
aAPC: Artificial antigen-presenting cell
CAR: Chimeric antigen receptor
CBI: Checkpoint-blockade Immunotherapy
CIK: Cytokine-induced killer
CML: Chronic myelogenous leukemia
CR: Complete regression
CTL: Cytotoxic T lymphocyte
CTLA-4: Cytotoxic T-lymphocyte-associated protein 4
Cy: Cyclophosphamide
Flu: Fludarabine
DAP10: Disulfide adaptor protein 10
DC: Dendritic cell
DFS: Disease-free survival
DLI: Donor lymphocyte infusion
FDA: Food and Drug Administration
GMP: Good manufacturing practice
GvHD: Graft versus host disease
GvT: Graft versus tumor
HLA: Human leukocyte antigen
HPC: Hematopoietic progenitor cells
HSC: Hematopoietic stem cells
HSCT: Hematopoietic stem-cell transplantation
IFN: Interferon
IL: Interleukin
KIR: Killer-cell immunoglobulin-like receptors
KIR-L: Killer-cell immunoglobulin-like receptors - ligand
LAK: Lymphokine-activated killer
MHC: Major histocompatibility complex
MNC: Mononuclear cell
NK: Natural Killer
NKG2D: Natural killer group 2D
NKT: Natural killer T cell
OR: Objective response
OSR: Overall survival rate
PD: Progressive disease
PD-1: Program cell death protein 1
PD-L1: Programmed death-ligand 1
PFS: Progression-free survival
PR: Partial response
QOL: Quality of life
RR: Response rate
SD: Stable disease
TAA: Tumour-associated antigen
TBI: Total body irradiation
TCR: T cell receptor
TIL: Tumor-infiltrating lymphocyte
UCB: Umbilical cord blood
Acknowledgments
None.
Author’s contributions
All authors equally contributed to this work. All authors read and approved the final manuscript.
Funding
This research is funded by Vietnam National University Ho Chi Minh City (VNU-HCM) under grant number C2020-18-26.
Availability of data and materials
Not applicable.
Ethics approval and consent to participate
Not applicable.
Consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing interests.
References
-
McCarthy EF. The toxins of William B. Coley and the treatment of bone and soft-tissue sarcomas. The Iowa orthopaedic journal.
2006;
26
:
154-158
.
-
Hinrichs CS, Rosenberg SA. Exploiting the curative potential of adoptive T‐cell therapy for cancer. Immunological reviews. 2014;257(1):56-71.
.
View Article PubMed Google Scholar -
Mellman I, Coukos G, Dranoff G. Cancer immunotherapy comes of age. Nature. 2011;480(7378):480-9.
.
View Article PubMed Google Scholar -
Mitchison NA. Passive transfer of transplantation immunity. Proceedings of the Royal Society of London Series B-Biological Sciences. 1954;142(906):72-87.
.
View Article PubMed Google Scholar -
Rosenberg SA, Restifo NP, Yang JC, Morgan RA, Dudley ME. Adoptive cell transfer: a clinical path to effective cancer immunotherapy. Nature Reviews Cancer. 2008;8(4):299-308.
.
View Article PubMed Google Scholar -
Dudley ME, Wunderlich JR, Robbins PF, Yang JC, Hwu P, Schwartzentruber DJ, et al. Cancer regression and autoimmunity in patients after clonal repopulation with antitumor lymphocytes. Science. 2002;298(5594):850-4 .
.
View Article PubMed Google Scholar -
Lindenberg MA, Retèl VP, van den Berg JH, Foppen MHG, Haanen JB, Van Harten WH. Treatment with tumor-infiltrating lymphocytes in advanced melanoma: evaluation of early clinical implementation of an advanced therapy medicinal product. Journal of immunotherapy (Hagerstown, Md: 1997). 2018;41(9):413 .
.
View Article PubMed Google Scholar -
Pagès F, Galon J, Dieu-Nosjean MC, Tartour E, Sautès-Fridman C, Fridman WH. Immune infiltration in human tumors: a prognostic factor that should not be ignored. Oncogene. 2010;29(8):1093-102 .
.
View Article PubMed Google Scholar -
Ogino S, Galon J, Fuchs CS, Dranoff G. Cancer immunology-analysis of host and tumor factors for personalized medicine. Nature Reviews Clinical Oncology. 2011;8(12):711-9.
.
View Article PubMed Google Scholar -
Burton AL, Roach BA, Mays MP, Chen AF, Ginter BAR, Vierling AM, et al. Prognostic Significance of Tumor Infiltrating Lymphocytes in Melanoma. The American Surgeon. 2011;77(2):188-92.
.
View Article PubMed Google Scholar -
Rosenberg SA, Lotze MT, Yang JC, Topalian SL, Chang AE, Schwartzentruber DJ, et al. Prospective Randomized Trial of High-Dose Interleukin-2 Alone or in Conjunction With Lymphokine-Activated Killer Cells for the Treatment of Patients With Advanced Cancer. JNCI: Journal of the National Cancer Institute. 1993;85(8):622-32.
.
View Article PubMed Google Scholar -
Farkona S, Diamandis EP, Blasutig IM. Cancer immunotherapy: the beginning of the end of cancer? BMC Medicine. 2016;14(1):73.
.
View Article PubMed Google Scholar -
Rosenberg SA, Spiess P, Lafreniere R. A new approach to the adoptive immunotherapy of cancer with tumor-infiltrating lymphocytes. Science. 1986;233(4770):1318.
.
View Article PubMed Google Scholar -
Fridman WH, Zitvogel L, Sautès-Fridman C, Kroemer G. The immune contexture in cancer prognosis and treatment. Nature Reviews Clinical Oncology. 2017;14(12):717-34.
.
View Article PubMed Google Scholar -
Rohaan MW, Wilgenhof S, Haanen JBAG. Adoptive cellular therapies: the current landscape. Virchows Arch. 2019;474(4):449-61.
.
View Article PubMed Google Scholar -
Robb RJ, Kutny RM, Chowdhry V. Purification and partial sequence analysis of human T-cell growth factor. Proc Natl Acad Sci U S A. 1983;80(19):5990-4.
.
View Article PubMed Google Scholar -
Topalian SL, Solomon D, Avis FP, Chang AE, Freerksen DL, Linehan WM, et al. Immunotherapy of patients with advanced cancer using tumor-infiltrating lymphocytes and recombinant interleukin-2: a pilot study. Journal of Clinical Oncology. 1988;6(5):839-53.
.
View Article PubMed Google Scholar -
Rosenberg SA, Packard BS, Aebersold PM, Solomon D, Topalian SL, Toy ST, et al. Use of Tumor-Infiltrating Lymphocytes and Interleukin-2 in the Immunotherapy of Patients with Metastatic Melanoma. New England Journal of Medicine. 1988;319(25):1676-80.
.
View Article PubMed Google Scholar -
Rosenberg SA, Yang JC, Sherry RM, Kammula US, Hughes MS, Phan GQ, et al. Durable Complete Responses in Heavily Pretreated Patients with Metastatic Melanoma Using T-Cell Transfer Immunotherapy. Clinical Cancer Research. 2011;17(13):4550 .
.
View Article PubMed Google Scholar -
Dafni U, Michielin O, Lluesma SM, Tsourti Z, Polydoropoulou V, Karlis D, et al. Efficacy of adoptive therapy with tumor-infiltrating lymphocytes and recombinant interleukin-2 in advanced cutaneous melanoma: a systematic review and meta-analysis. Annals of Oncology. 2019;30(12):1902-13.
.
View Article PubMed Google Scholar -
Forget M-A, Haymaker C, Hess KR, Meng YJ, Creasy C, Karpinets T, et al. Prospective Analysis of Adoptive TIL Therapy in Patients with Metastatic Melanoma: Response, Impact of Anti-CTLA4, and Biomarkers to Predict Clinical Outcome. Clinical Cancer Research. 2018;24(18):4416.
.
View Article PubMed Google Scholar -
Mehta GU, Malekzadeh P, Shelton T, White DE, Butman JA, Yang JC, et al. Outcomes of Adoptive Cell Transfer With Tumor-infiltrating Lymphocytes for Metastatic Melanoma Patients With and Without Brain Metastases. J Immunother. 2018;41(5):241-7.
.
View Article PubMed Google Scholar -
Stevanović S, Helman SR, Wunderlich JR, Langhan MM, Doran SL, Kwong MLM, et al. A Phase II Study of Tumor-infiltrating Lymphocyte Therapy for Human Papillomavirus-associated Epithelial Cancers. Clinical Cancer Research. 2019;25(5):1486.
.
View Article PubMed Google Scholar -
Li J, Chen Q-Y, He J, Li Z-L, Tang X-F, Chen S-P, et al. Phase I trial of adoptively transferred tumor-infiltrating lymphocyte immunotherapy following concurrent chemoradiotherapy in patients with locoregionally advanced nasopharyngeal carcinoma. OncoImmunology. 2015;4(2):e976507.
.
View Article PubMed Google Scholar -
Zacharakis N, Chinnasamy H, Black M, Xu H, Lu Y-C, Zheng Z, et al. Immune recognition of somatic mutations leading to complete durable regression in metastatic breast cancer. Nat Med. 2018;24(6):724-30.
.
View Article PubMed Google Scholar -
Wolf B, Zimmermann S, Arber C, Irving M, Trueb L, Coukos G. Safety and Tolerability of Adoptive Cell Therapy in Cancer. Drug Safety. 2019;42(2):315-34.
.
View Article PubMed Google Scholar -
Tao Z, Li S, Ichim TE, Yang J, Riordan N, Yenugonda V, et al. Cellular immunotherapy of cancer: an overview and future directions. Immunotherapy. 2017;9(7):589-606.
.
View Article PubMed Google Scholar -
Kiessling R, Klein E, Wigzell H. "Natural"︁ killer cells in the mouse. I. Cytotoxic cells with specificity for mouse Moloney leukemia cells. Specificity and distribution according to genotype. European Journal of Immunology. 1975;5(2):112-7.
.
View Article PubMed Google Scholar -
Herberman RB, Nunn ME, Lavrin DH. Natural cytotoxic reactivity of mouse lymphoid cells against syngeneic and allogeneic tumors. I. Distribution of reactivity and specificity. International Journal of Cancer. 1975;16(2):216-29.
.
View Article PubMed Google Scholar -
Orange JS. Natural killer cell deficiency. Journal of Allergy and Clinical Immunology. 2013;132(3):515-25.
.
View Article PubMed Google Scholar -
Remark R, Alifano M, Cremer I, Lupo A, Dieu-Nosjean M-C, Riquet M, et al. Characteristics and Clinical Impacts of the Immune Environments in Colorectal and Renal Cell Carcinoma Lung Metastases: Influence of Tumor Origin. Clinical Cancer Research. 2013;19(15):4079.
.
View Article PubMed Google Scholar -
Albertsson PA, Basse PH, Hokland M, Goldfarb RH, Nagelkerke JF, Nannmark U, et al. NK cells and the tumour microenvironment: implications for NK-cell function and anti-tumour activity. Trends in Immunology. 2003;24(11):603-9.
.
View Article PubMed Google Scholar -
Mandal R, Şenbabaoğlu Y, Desrichard A, Havel JJ, Dalin MG, Riaz N, et al. The head and neck cancer immune landscape and its immunotherapeutic implications. JCI Insight. 2016;1(17).
.
View Article PubMed Google Scholar -
Campbell KS, Hasegawa J. Natural killer cell biology: An update and future directions. Journal of Allergy and Clinical Immunology. 2013;132(3):536-44.
.
View Article PubMed Google Scholar -
Hu W, Wang G, Huang D, Sui M, Xu Y. Cancer Immunotherapy Based on Natural Killer Cells: Current Progress and New Opportunities. 2019;10(1205).
.
View Article PubMed Google Scholar -
Pegram HJ, Andrews DM, Smyth MJ, Darcy PK, Kershaw MH. Activating and inhibitory receptors of natural killer cells. 2011;89(2):216-24. https://doi.org/10.1038/icb.2010.78
.
PubMed Google Scholar -
Morvan MG, Lanier LL. NK cells and cancer: you can teach innate cells new tricks. Nature Reviews Cancer. 2016;16(1):7-19.
.
View Article PubMed Google Scholar -
Long EO, Kim HS, Liu D, Peterson ME, Rajagopalan S. Controlling natural killer cell responses: integration of signals for activation and inhibition. Annu Rev Immunol. 2013;31:227-58.
.
View Article PubMed Google Scholar -
Ljunggren HG, Kärre K. In search of the 'missing self': MHC molecules and NK cell recognition. Immunol Today.
1990;
11
(7)
:
237-244
.
View Article PubMed Google Scholar -
Veluchamy JP, Kok N, van der Vliet HJ, Verheul HMW, de Gruijl TD, Spanholtz J. The Rise of Allogeneic Natural Killer Cells As a Platform for Cancer Immunotherapy: Recent Innovations and Future Developments. Front Immunol.
2017;
31
(8)
:
631
.
View Article PubMed Google Scholar -
Lupo KB, Matosevic S. Natural Killer Cells as Allogeneic Effectors in Adoptive Cancer Immunotherapy. Cancers.
2019;
11
(6)
:
769
.
View Article Google Scholar -
Burns LJ, Weisdorf DJ, DeFor TE, Vesole DH, Repka TL, Blazar BR, et al. IL-2-based immunotherapy after autologous transplantation for lymphoma and breast cancer induces immune activation and cytokine release: a phase I/II trial. Bone Marrow Transplantation. 2003;32(2):177-86.
.
View Article PubMed Google Scholar -
Lister J, Rybka WB, Donnenberg AD, deMagalhaes-Silverman M, Pincus SM, Bloom EJ, et al. Autologous peripheral blood stem cell transplantation and adoptive immunotherapy with activated natural killer cells in the immediate posttransplant period. Clinical Cancer Research. 1995;1(6):607.
.
-
Terme M, Ullrich E, Delahaye NF, Chaput N, Zitvogel L. Natural killer cell-directed therapies: moving from unexpected results to successful strategies. Nature Immunology. 2008;9(5):486-94.
.
View Article PubMed Google Scholar -
Parkhurst MR, Riley JP, Dudley ME, Rosenberg SA. Adoptive Transfer of Autologous Natural Killer Cells Leads to High Levels of Circulating Natural Killer Cells but Does Not Mediate Tumor Regression. Clinical Cancer Research. 2011;17(19):6287.
.
View Article PubMed Google Scholar -
Ljunggren H-G, Kärre K. In search of the 'missing self': MHC molecules and NK cell recognition. Immunology Today. 1990;
11
:
237-244
.
View Article Google Scholar -
Miller JS, Rooney CM, Curtsinger J, McElmurry R, McCullar V, Verneris MR, et al. Expansion and Homing of Adoptively Transferred Human Natural Killer Cells in Immunodeficient Mice Varies with Product Preparation and In Vivo Cytokine Administration: Implications for Clinical Therapy. Biology of Blood and Marrow Transplantation. 2014;20(8):1252-7.
.
View Article PubMed Google Scholar -
Pittari G, Filippini P, Gentilcore G, Grivel J-C, Rutella S. Revving up Natural Killer Cells and Cytokine-Induced Killer Cells Against Hematological Malignancies. Front Immunol. 2015;6:230-.
.
View Article PubMed Google Scholar -
Krause SW, Gastpar R, Andreesen R, Gross C, Ullrich H, Thonigs G, et al. Treatment of Colon and Lung Cancer Patients with ex Vivo Heat Shock Protein 70-Peptide-Activated, Autologous Natural Killer Cells. Clinical Cancer Research. 2004;10(11):3699.
.
View Article PubMed Google Scholar -
Ishikawa E, Tsuboi K, Saijo K, Harada H, Takano S, Nose T, et al. Autologous Natural Killer Cell Therapy for Human Recurrent Malignant Glioma. Anticancer Research. 2004;24(3B):1861-71.
.
-
Li L, Li W, Wang C, Yan X, Wang Y, Niu C, et al. Adoptive transfer of natural killer cells in combination with chemotherapy improves outcomes of patients with locally advanced colon carcinoma. Cytotherapy. 2018;20(1):134-48.
.
View Article PubMed Google Scholar -
Kokowski K, Stangl S, Seier S, Hildebrandt M, Vaupel P, Multhoff G. Radiochemotherapy combined with NK cell transfer followed by second-line PD-1 inhibition in a patient with NSCLC stage IIIb inducing long-term tumor control: a case study. Strahlentherapie und Onkologie. 2019;195(4):352-61.
.
View Article PubMed Google Scholar -
Lupo KB, Matosevic S. Natural Killer Cells as Allogeneic Effectors in Adoptive Cancer Immunotherapy. Cancers (Basel). 2019;11(6):769.
.
View Article PubMed Google Scholar -
Ruggeri L, Capanni M, Casucci M, Volpi I, Tosti A, Perruccio K, et al. Role of natural killer cell alloreactivity in HLA-mismatched hematopoietic stem cell transplantation. Blood. 1999;94(1):333-9.
.
View Article PubMed Google Scholar -
Ruggeri L, Capanni M, Urbani E, Perruccio K, Shlomchik WD, Tosti A, et al. Effectiveness of Donor Natural Killer Cell Alloreactivity in Mismatched Hematopoietic Transplants. Science. 2002;295(5562):2097.
.
View Article PubMed Google Scholar -
Ruggeri L, Parisi S, Urbani E, Curti A. Alloreactive Natural Killer Cells for the Treatment of Acute Myeloid Leukemia: From Stem Cell Transplantation to Adoptive Immunotherapy. 2015;6(479).
.
View Article PubMed Google Scholar -
Miller JS, Soignier Y, Panoskaltsis-Mortari A, McNearney SA, Yun GH, Fautsch SK, et al. Successful adoptive transfer and in vivo expansion of human haploidentical NK cells in patients with cancer. Blood. 2005;105(8):3051-7.
.
View Article PubMed Google Scholar -
Björklund
Andreas T,
Carlsten
Mattias,
Sohlberg
Ebba,
Liu
Lisa L,
Clancy
Trevor,
Karimi
Mohsen,
Cooley
Sarah,
Miller
Jeffrey S,
Klimkowska
Monika,
Schaffer
Marie,
Complete remission with reduction of high-risk clones following haploidentical NK-cell therapy against MDS and AML. Clinical Cancer Research.
2018;
24
(8)
:
1834-1844
.
View Article PubMed Google Scholar -
Curti A, Ruggeri L, D'Addio A, Bontadini A, Dan E, Motta MR, et al. Successful transfer of alloreactive haploidentical KIR ligand-mismatched natural killer cells after infusion in elderly high risk acute myeloid leukemia patients. Blood. 2011;118(12):3273-9.
.
View Article PubMed Google Scholar -
Bachanova V, Cooley S, Defor TE, Verneris MR, Zhang B, McKenna DH, et al. Clearance of acute myeloid leukemia by haploidentical natural killer cells is improved using IL-2 diphtheria toxin fusion protein. Blood. 2014;123(25):3855-63.
.
View Article PubMed Google Scholar -
Rubnitz JE, Inaba H, Ribeiro RC, Pounds S, Rooney B, Bell T, et al. NKAML: a pilot study to determine the safety and feasibility of haploidentical natural killer cell transplantation in childhood acute myeloid leukemia. J Clin Oncol. 2010;28(6):955-9.
.
View Article PubMed Google Scholar -
Yang Y, Lim O, Kim TM, Ahn Y-O, Choi H, Chung H, et al. Phase I Study of Random Healthy Donor-Derived Allogeneic Natural Killer Cell Therapy in Patients with Malignant Lymphoma or Advanced Solid Tumors. Cancer Immunology Research. 2016;4(3):215.
.
View Article PubMed Google Scholar -
Iliopoulou EG, Kountourakis P, Karamouzis MV, Doufexis D, Ardavanis A, Baxevanis CN, et al. A phase I trial of adoptive transfer of allogeneic natural killer cells in patients with advanced non-small cell lung cancer. Cancer Immunology, Immunotherapy. 2010;59(12):1781-9.
.
View Article PubMed Google Scholar -
Geller MA, Cooley S, Judson PL, Ghebre R, Carson LF, Argenta PA, et al. A phase II study of allogeneic natural killer cell therapy to treat patients with recurrent ovarian and breast cancer. Cytotherapy. 2011;13(1):98-107.
.
View Article PubMed Google Scholar -
Dolstra H, Roeven MWH, Spanholtz J, Hangalapura BN, Tordoir M, Maas F, et al. Successful Transfer of Umbilical Cord Blood CD34+ Hematopoietic Stem and Progenitor-derived NK Cells in Older Acute Myeloid Leukemia Patients. Clinical Cancer Research. 2017;23(15):4107.
.
View Article PubMed Google Scholar -
Tomchuck
Suzanne L,
Leung
Wing H,
Dallas
Mari H,
Enhanced cytotoxic function of natural killer and CD3+ CD56+ cells in cord blood after culture. Biology of Blood and Marrow Transplantation.
2015;
21
(1)
:
39-49
.
View Article PubMed Google Scholar -
Nomura A, Takada H, Jin C-H, Tanaka T, Ohga S, Hara T. Functional analyses of cord blood natural killer cells and T cells: a distinctive interleukin-18 response. Experimental Hematology. 2001;29(10):1169-76.
.
View Article Google Scholar -
Shah N, Martin-Antonio B, Yang H, Ku S, Lee DA, Cooper LJN, et al. Antigen Presenting Cell-Mediated Expansion of Human Umbilical Cord Blood Yields Log-Scale Expansion of Natural Killer Cells with Anti-Myeloma Activity. PLOS ONE. 2013;8(10):e76781.
.
View Article PubMed Google Scholar -
Ayello J, Hochberg J, Flower A, Chu Y, Baxi LV, Quish W, et al. Genetically re-engineered K562 cells significantly expand and functionally activate cord blood natural killer cells: Potential for adoptive cellular immunotherapy. Experimental Hematology. 2017;46:38-47.
.
View Article PubMed Google Scholar -
Vasu S, Berg M, Davidson-Moncada J, Tian X, Cullis H, Childs RW. A novel method to expand large numbers of CD56+ natural killer cells from a minute fraction of selectively accessed cryopreserved cord blood for immunotherapy after transplantation. Cytotherapy. 2015;17(11):1582-93.
.
View Article PubMed Google Scholar -
Sarvaria A, Jawdat D, Madrigal JA, Saudemont A. Umbilical Cord Blood Natural Killer Cells, Their Characteristics, and Potential Clinical Applications. Front Immunol. 2017;8:329.
.
View Article PubMed Google Scholar -
Shah N, Li L, McCarty J, Kaur I, Yvon E, Shaim H, et al. Phase I study of cord blood-derived natural killer cells combined with autologous stem cell transplantation in multiple myeloma. Br J Haematol. 2017;177(3):457-66.
.
View Article PubMed Google Scholar -
Veluchamy JP, Lopez-Lastra S, Spanholtz J, Bohme F, Kok N, Heideman DAM, et al. In Vivo Efficacy of Umbilical Cord Blood Stem Cell-Derived NK Cells in the Treatment of Metastatic Colorectal Cancer. Front Immunol. 2017;8:87-.
.
View Article PubMed Google Scholar -
Cany J, van der Waart AB, Tordoir M, Franssen GM, Hangalapura BN, de Vries J, et al. Natural Killer Cells Generated from Cord Blood Hematopoietic Progenitor Cells Efficiently Target Bone Marrow-Residing Human Leukemia Cells in NOD/SCID/IL2Rgnull Mice. PLOS ONE. 2013;8(6):e64384.
.
View Article PubMed Google Scholar -
Cany J, van der Waart AB, Spanholtz J, Tordoir M, Jansen JH, van der Voort R, et al. Combined IL-15 and IL-12 drives the generation of CD34+-derived natural killer cells with superior maturation and alloreactivity potential following adoptive transfer. OncoImmunology. 2015;4(7):e1017701.
.
View Article PubMed Google Scholar -
Spanholtz J, Preijers F, Tordoir M, Trilsbeek C, Paardekooper J, de Witte T, et al. Clinical-Grade Generation of Active NK Cells from Cord Blood Hematopoietic Progenitor Cells for Immunotherapy Using a Closed-System Culture Process. PLOS ONE. 2011;6(6):e20740.
.
View Article PubMed Google Scholar -
Leung W. Infusions of allogeneic natural killer cells as cancer therapy. Clinical cancer research : an official journal of the American Association for Cancer Research. 2014;20(13):3390-400.
.
View Article PubMed Google Scholar -
Tarn YK, Martinson JA, Doligosa K, Klingernann HG. Ex vivo expansion of the highly cytotoxic human natural killer cell line NK-92 under current good manufacturing practice conditions for clinical adoptive cellular immunotherapy. Cytotherapy. 2003;5(3):259-72.
.
View Article PubMed Google Scholar -
Klingemann HG, Wong E, Maki G. A cytotoxic NK-cell line (NK-92) for ex vivo purging of leukemia from blood. Biology of blood and marrow transplantation : journal of the American Society for Blood and Marrow Transplantation. 1996;2(2):68-75.
.
-
Klingemann H, Boissel L, Toneguzzo F. Natural Killer Cells for Immunotherapy - Advantages of the NK-92 Cell Line over Blood NK Cells. Front Immunol. 2016;7:91.
.
View Article PubMed Google Scholar -
Maki G, Klingemann HG, Martinson JA, Tam YK. Factors regulating the cytotoxic activity of the human natural killer cell line, NK-92. Journal of hematotherapy & stem cell research. 2001;10(3):369-83.
.
View Article PubMed Google Scholar -
Suck G, Odendahl M, Nowakowska P, Seidl C, Wels WS, Klingemann HG, et al. NK-92: an 'off-the-shelf therapeutic' for adoptive natural killer cell-based cancer immunotherapy. Cancer immunology, immunotherapy : CII. 2016;65(4):485-92.
.
View Article PubMed Google Scholar -
Tonn T, Seifried E. Natural Killer Cells for the Treatment of Malignancies*. Transfusion Medicine and Hemotherapy. 2006;33(2):144-9.
.
View Article Google Scholar -
Arai S, Meagher R, Swearingen M, Myint H, Rich E, Martinson J, et al. Infusion of the allogeneic cell line NK-92 in patients with advanced renal cell cancer or melanoma: a phase I trial. Cytotherapy. 2008;10(6):625-32.
.
View Article PubMed Google Scholar -
Williams BA, Law AD, Routy B, denHollander N, Gupta V, Wang XH, et al. A phase I trial of NK-92 cells for refractory hematological malignancies relapsing after autologous hematopoietic cell transplantation shows safety and evidence of efficacy. Oncotarget. 2017;8(51):89256-68.
.
View Article PubMed Google Scholar -
Boyiadzis M, Agha M, Redner RL, Sehgal A, Im A, Hou JZ, et al. Phase 1 clinical trial of adoptive immunotherapy using "off-the-shelf" activated natural killer cells in patients with refractory and relapsed acute myeloid leukemia. Cytotherapy. 2017;19(10):1225-32.
.
View Article PubMed Google Scholar -
Tonn T, Schwabe D, Klingemann HG, Becker S, Esser R, Koehl U, et al. Treatment of patients with advanced cancer with the natural killer cell line NK-92. Cytotherapy. 2013;15(12):1563-70.
.
View Article PubMed Google Scholar -
Matosevic S. Viral and Nonviral Engineering of Natural Killer Cells as Emerging Adoptive Cancer Immunotherapies. Journal of immunology research. 2018;2018:4054815.
.
View Article PubMed Google Scholar -
Schmidt-Wolf IG, Lefterova P, Mehta BA, Fernandez LP, Huhn D, Blume KG, et al. Phenotypic characterization and identification of effector cells involved in tumor cell recognition of cytokine-induced killer cells. Exp Hematol. 1993;21(13):1673-9.
.
-
Mata-Molanes JJ, Sureda González M, Valenzuela Jiménez B, Martínez Navarro EM, Brugarolas Masllorens A. Cancer Immunotherapy with Cytokine-Induced Killer Cells. Targeted oncology. 2017;12(3):289-99.
.
View Article PubMed Google Scholar -
Linn YC, Lau SKJ, Liu BH, Ng LH, Yong HX, Hui KM. Characterization of the recognition and functional heterogeneity exhibited by cytokine-induced killer cell subsets against acute myeloid leukaemia target cell. Immunology. 2009;126(3):423-35.
.
View Article PubMed Google Scholar -
Rettinger E, Kuçi S, Naumann I, Becker P, Kreyenberg H, Anzaghe M, et al. The cytotoxic potential of interleukin-15-stimulated cytokine-induced killer cells against leukemia cells. Cytotherapy. 2012;14(1):91-103.
.
View Article PubMed Google Scholar -
Iudicone P, Fioravanti D, Cicchetti E, Zizzari IG, Pandolfi A, Scocchera R, et al. Interleukin-15 enhances cytokine induced killer (CIK) cytotoxic potential against epithelial cancer cell lines via an innate pathway. Human immunology. 2016;77(12):1239-47.
.
View Article PubMed Google Scholar -
Rettinger E, Huenecke S, Bonig H, Merker M, Jarisch A, Soerensen J, et al. Interleukin-15-activated cytokine-induced killer cells may sustain remission in leukemia patients after allogeneic stem cell transplantation: feasibility, safety and first insights on efficacy. Haematologica. 2016;101(4):e153-e6.
.
View Article PubMed Google Scholar -
Bremm M, Pfeffermann L-M, Cappel C, Katzki V, Erben S, Betz S, et al. Improving Clinical Manufacturing of IL-15 Activated Cytokine-Induced Killer (CIK) Cells. Front Immunol. 2019;10:1218-.
.
View Article PubMed Google Scholar -
Mehta BA, Schmidt-Wolf IG, Weissman IL, Negrin RS. Two pathways of exocytosis of cytoplasmic granule contents and target cell killing by cytokine-induced CD3+ CD56+ killer cells. Blood. 1995;86(9):3493-9.
.
View Article PubMed Google Scholar -
Linn YC, Wang SM, Hui KM. Comparative gene expression profiling of cytokine-induced killer cells in response to acute myloid leukemic and acute lymphoblastic leukemic stimulators using oligonucleotide arrays. Exp Hematol. 2005;33(6):671-81.
.
View Article PubMed Google Scholar -
Lee HK, Kim YG, Kim JS, Park EJ, Kim B, Park KH, et al. Cytokine-induced killer cells interact with tumor lysate-pulsed dendritic cells via CCR5 signaling. Cancer letters. 2016;378(2):142-9.
.
View Article PubMed Google Scholar -
Introna M, Correnti F. Innovative Clinical Perspectives for CIK Cells in Cancer Patients. Int J Mol Sci. 2018;19(2):358.
.
View Article PubMed Google Scholar -
Pievani A, Borleri G, Pende D, Moretta L, Rambaldi A, Golay J, et al. Dual-functional capability of CD3+CD56+ CIK cells, a T-cell subset that acquires NK function and retains TCR-mediated specific cytotoxicity. Blood. 2011;118(12):3301-10.
.
View Article PubMed Google Scholar -
Tang Q, Grzywacz B, Wang H, Kataria N, Cao Q, Wagner JE, et al. Umbilical cord blood T cells express multiple natural cytotoxicity receptors after IL-15 stimulation, but only NKp30 is functional. J Immunol. 2008;181(7):4507-15.
.
View Article PubMed Google Scholar -
Gao X, Mi Y, Guo N, Xu H, Xu L, Gou X, et al. Cytokine-Induced Killer Cells As Pharmacological Tools for Cancer Immunotherapy. Front Immunol. 2017;8:774-.
.
View Article PubMed Google Scholar -
Thanendrarajan S, Nowak M, Abken H, Schmidt-Wolf IG. Combining cytokine-induced killer cells with vaccination in cancer immunotherapy: more than one plus one? Leukemia research. 2011;35(9):1136-42.
.
View Article PubMed Google Scholar -
Mesiano G, Grignani G, Fiorino E, Leuci V, Rotolo R, D'Ambrosio L, et al. Cytokine Induced Killer cells are effective against sarcoma cancer stem cells spared by chemotherapy and target therapy. Oncoimmunology. 2018;7(11):e1465161-e.
.
View Article PubMed Google Scholar -
Gammaitoni L, Giraudo L, Macagno M, Leuci V, Mesiano G, Rotolo R, et al. Cytokine-Induced Killer Cells Kill Chemo-surviving Melanoma Cancer Stem Cells. Clinical cancer research : an official journal of the American Association for Cancer Research. 2017;23(9):2277-88.
.
View Article PubMed Google Scholar -
Yang T, Zhang W, Wang L, Xiao C, Wang L, Gong Y, et al. Co-culture of dendritic cells and cytokine-induced killer cells effectively suppresses liver cancer stem cell growth by inhibiting pathways in the immune system. BMC cancer. 2018;18(1):984.
.
View Article PubMed Google Scholar -
Schmidt-Wolf IG, Negrin RS, Kiem HP, Blume KG, Weissman IL. Use of a SCID mouse/human lymphoma model to evaluate cytokine-induced killer cells with potent antitumor cell activity. The Journal of experimental medicine. 1991;174(1):139-49.
.
View Article PubMed Google Scholar -
Edinger M, Cao YA, Verneris MR, Bachmann MH, Contag CH, Negrin RS. Revealing lymphoma growth and the efficacy of immune cell therapies using in vivo bioluminescence imaging. Blood. 2003;101(2):640-8.
.
View Article PubMed Google Scholar -
Schmidt-Wolf IG, Finke S, Trojaneck B, Denkena A, Lefterova P, Schwella N, et al. Phase I clinical study applying autologous immunological effector cells transfected with the interleukin-2 gene in patients with metastatic renal cancer, colorectal cancer and lymphoma. Br J Cancer. 1999;81(6):1009-16.
.
View Article PubMed Google Scholar -
Leemhuis T, Wells S, Scheffold C, Edinger M, Negrin RS. A phase I trial of autologous cytokine-induced killer cells for the treatment of relapsed Hodgkin disease and non-Hodgkin lymphoma. Biology of blood and marrow transplantation : journal of the American Society for Blood and Marrow Transplantation. 2005;11(3):181-7.
.
View Article PubMed Google Scholar -
Lee JH, Lee JH, Lim YS, Yeon JE, Song TJ, Yu SJ, et al. Adjuvant immunotherapy with autologous cytokine-induced killer cells for hepatocellular carcinoma. Gastroenterology. 2015;148(7):1383-91.e6.
.
View Article PubMed Google Scholar -
Kong D-S, Nam D-H, Kang S-H, Lee JW, Chang J-H, Kim J-H, et al. Phase III randomized trial of autologous cytokine-induced killer cell immunotherapy for newly diagnosed glioblastoma in Korea. Oncotarget. 2017;8(4):7003-13.
.
View Article PubMed Google Scholar -
Li X, Dai X, Shi L, Jiang Y, Chen X, Chen L, et al. Phase II/III Study of Radiofrequency Ablation Combined with Cytokine-Induced Killer Cells Treating Colorectal Liver Metastases. Cellular physiology and biochemistry : international journal of experimental cellular physiology, biochemistry, and pharmacology. 2016;40(1-2):137-45.
.
View Article PubMed Google Scholar -
Schmeel FC, Schmeel LC, Gast S-M, Schmidt-Wolf IGH. Adoptive immunotherapy strategies with cytokine-induced killer (CIK) cells in the treatment of hematological malignancies. Int J Mol Sci. 2014;15(8):14632-48.
.
View Article PubMed Google Scholar -
Schmeel LC, Schmeel FC, Coch C, Schmidt-Wolf IG. Cytokine-induced killer (CIK) cells in cancer immunotherapy: report of the international registry on CIK cells (IRCC). Journal of cancer research and clinical oncology. 2015;141(5):839-49.
.
View Article PubMed Google Scholar -
Li XD, Xu B, Wu J, Ji M, Xu BH, Jiang JT, et al. Review of Chinese clinical trials on CIK cell treatment for malignancies. Clinical & translational oncology : official publication of the Federation of Spanish Oncology Societies and of the National Cancer Institute of Mexico. 2012;14(2):102-8.
.
View Article Google Scholar -
Olioso P, Giancola R, Di Riti M, Contento A, Accorsi P, Iacone A. Immunotherapy with cytokine induced killer cells in solid and hematopoietic tumours: a pilot clinical trial. Hematological oncology. 2009;27(3):130-9.
.
View Article PubMed Google Scholar -
Sangiolo D, Martinuzzi E, Todorovic M, Vitaggio K, Vallario A, Jordaney N, et al. Alloreactivity and anti-tumor activity segregate within two distinct subsets of cytokine-induced killer (CIK) cells: implications for their infusion across major HLA barriers. International immunology. 2008;20(7):841-8.
.
View Article PubMed Google Scholar -
Todorovic M, Mesiano G, Gammaitoni L, Leuci V, Giraudo Diego L, Cammarata C, et al. Ex vivo allogeneic stimulation significantly improves expansion of cytokine-induced killer cells without increasing their alloreactivity across HLA barriers. J Immunother. 2012;35(7):579-86.
.
View Article PubMed Google Scholar -
Kuçi S, Rettinger E, Voss B, Weber G, Stais M, Kreyenberg H, et al. Efficient lysis of rhabdomyosarcoma cells by cytokine-induced killer cells: implications for adoptive immunotherapy after allogeneic stem cell transplantation. Haematologica. 2010;95(9):1579-86.
.
View Article PubMed Google Scholar -
Nishimura R, Baker J, Beilhack A, Zeiser R, Olson JA, Sega EI, et al. In vivo trafficking and survival of cytokine-induced killer cells resulting in minimal GVHD with retention of antitumor activity. Blood. 2008;112(6):2563-74.
.
View Article PubMed Google Scholar -
Baker J, Verneris MR, Ito M, Shizuru JA, Negrin RS. Expansion of cytolytic CD8+ natural killer T cells with limited capacity for graft-versus-host disease induction due to interferon γ production. Blood. 2001;97(10):2923-31.
.
View Article PubMed Google Scholar -
Rettinger E, Kreyenberg H, Merker M, Kuçi S, Willasch A, Bug G, et al. Immunomagnetic selection or irradiation eliminates alloreactive cells but also reduces anti-tumor potential of cytokine-induced killer cells: implications for unmanipulated cytokine-induced killer cell infusion. Cytotherapy. 2014;16(6):835-44.
.
View Article PubMed Google Scholar -
Yang YG, Dey BR, Sergio JJ, Pearson DA, Sykes M. Donor-derived interferon gamma is required for inhibition of acute graft-versus-host disease by interleukin 12. J Clin Invest. 1998;102(12):2126-35.
.
View Article PubMed Google Scholar -
Introna M, Borleri G, Conti E, Franceschetti M, Barbui AM, Broady R, et al. Repeated infusions of donor-derived cytokine-induced killer cells in patients relapsing after allogeneic stem cell transplantation: a phase I study. Haematologica. 2007;92(7):952-9.
.
View Article PubMed Google Scholar -
Introna M, Lussana F, Algarotti A, Gotti E, Valgardsdottir R, Micò C, et al. Phase II Study of Sequential Infusion of Donor Lymphocyte Infusion and Cytokine-Induced Killer Cells for Patients Relapsed after Allogeneic Hematopoietic Stem Cell Transplantation. Biology of blood and marrow transplantation : journal of the American Society for Blood and Marrow Transplantation. 2017;23(12):2070-2078.
.
View Article PubMed Google Scholar -
Linn YC, Niam M, Chu S, Choong A, Yong HX, Heng KK, et al. The anti-tumour activity of allogeneic cytokine-induced killer cells in patients who relapse after allogeneic transplant for haematological malignancies. Bone Marrow Transplant. 2012;47(7):957-66.
.
View Article PubMed Google Scholar -
Merker M, Salzmann-Manrique E, Katzki V, Huenecke S, Bremm M, Bakhtiar S, et al. Clearance of Hematologic Malignancies by Allogeneic Cytokine-Induced Killer Cell or Donor Lymphocyte Infusions. Biology of blood and marrow transplantation : journal of the American Society for Blood and Marrow Transplantation. 2019;25(7):1281-92.
.
View Article PubMed Google Scholar -
Wang S, Zhang H, Liu C, Jiao X, Liu D, Du W, et al. Human leukocyte antigen-haploidentical donor-derived cytokine-induced killer cells are safe and prolong the survival of patients with advanced non-small cell lung cancer. Oncology letters. 2014;8(6):2727-33.
.
View Article PubMed Google Scholar -
Zhang Z, Zhao X, Zhang T, Wang L, Yang L, Huang L, et al. Phenotypic characterization and anti-tumor effects of cytokine-induced killer cells derived from cord blood. Cytotherapy. 2015;17(1):86-97.
.
View Article PubMed Google Scholar -
Durrieu L, Lemieux W, Dieng MM, Fontaine F, Duval M, Le Deist F, et al. Implication of different effector mechanisms by cord blood-derived and peripheral blood-derived cytokine-induced killer cells to kill precursor B acute lymphoblastic leukemia cell lines. Cytotherapy. 2014;16(6):845-56.
.
View Article PubMed Google Scholar -
Introna M, Franceschetti M, Ciocca A, Borleri G, Conti E, Golay J, et al. Rapid and massive expansion of cord blood-derived cytokine-induced killer cells: an innovative proposal for the treatment of leukemia relapse after cord blood transplantation. Bone Marrow Transplant. 2006;38(9):621-7.
.
View Article PubMed Google Scholar -
Niu Q, Wang W, Li Y, Qin S, Wang Y, Wan G, et al. Cord blood-derived cytokine-induced killer cells biotherapy combined with second-line chemotherapy in the treatment of advanced solid malignancies. International immunopharmacology. 2011;11(4):449-56.
.
View Article PubMed Google Scholar -
Introna M, Pievani A, Borleri G, Capelli C, Algarotti A, Micò C, et al. Feasibility and safety of adoptive immunotherapy with CIK cells after cord blood transplantation. Biology of blood and marrow transplantation : journal of the American Society for Blood and Marrow Transplantation. 2010;16(11):1603-7.
.
View Article PubMed Google Scholar -
Zhou X, Zhu J, Sun H, Shao L, Xu M, Guo H. Family haploidentical donor-derived cytokine-induced killer cell biotherapy combined with bortezomib in two patients with relapsed multiple myeloma in a non-allogeneic transplant setting. Leukemia & lymphoma. 2013;54(1):209-11.
.
View Article PubMed Google Scholar -
Rettinger E, Bonig H, Wehner S, Lucchini G, Willasch A, Jarisch A, et al. Feasibility of IL-15-activated cytokine-induced killer cell infusions after haploidentical stem cell transplantation. Bone Marrow Transplantation. 2013;48(8):1141-3.
.
View Article PubMed Google Scholar -
Golay J, Martinelli S, Alzani R, Cribioli S, Albanese C, Gotti E, et al. Cord blood-derived cytokine-induced killer cells combined with blinatumomab as a therapeutic strategy for CD19(+) tumors. Cytotherapy. 2018;20(8):1077-88.
.
View Article PubMed Google Scholar -
Zhang Z, Wang L, Luo Z, Zhao X, Huang J, Li H, et al. Efficacy and safety of cord blood-derived cytokine-induced killer cells in treatment of patients with malignancies. Cytotherapy. 2015;17(8):1130-8.
.
View Article PubMed Google Scholar -
Wang L, Huang S, Dang Y, Li M, Bai W, Zhong Z, et al. Cord blood-derived cytokine-induced killer cellular therapy plus radiation therapy for esophageal cancer: a case report. Medicine. 2014;93(28):e340.
.
View Article PubMed Google Scholar -
Kumar BV, Connors TJ, Farber DL. Human T cell development, localization, and function throughout life. Immunity. 2018;48(2):202-13.
.
View Article PubMed Google Scholar -
Zhao L, Cao YJ. Engineered T Cell Therapy for Cancer in the Clinic. Front Immunol. 2019;10.
.
View Article PubMed Google Scholar -
Hedrick SM, Cohen DI, Nielsen EA, Davis MM. Isolation of cDNA clones encoding T cell-specific membrane-associated proteins. Nature. 1984;308(5955):149-53.
.
View Article PubMed Google Scholar -
Davis MM, Bjorkman PJ. T-cell antigen receptor genes and T-cell recognition. Nature. 1988;334(6181):395-402.
.
View Article PubMed Google Scholar -
Sundberg EJ, Deng L, Mariuzza RA, editors. TCR recognition of peptide/MHC class II complexes and superantigens. Seminars in immunology; 2007: Elsevier.
.
View Article PubMed Google Scholar -
Stone JD, Chervin AS, Kranz DM. T‐cell receptor binding affinities and kinetics: impact on T‐cell activity and specificity. Immunology. 2009;126(2):165-76.
.
View Article PubMed Google Scholar -
Dong D, Zheng L, Lin J, Zhang B, Zhu Y, Li N, et al. Structural basis of assembly of the human T cell receptor-CD3 complex. Nature. 2019;573(7775):546-52.
.
View Article PubMed Google Scholar -
Garber K. Driving T-cell immunotherapy to solid tumors. Nature Publishing Group; 2018.
.
View Article PubMed Google Scholar -
Zhang J, Wang L. The emerging world of TCR-T cell trials against cancer: a systematic review. Technology in cancer research & treatment. 2019;18:1533033819831068.
.
View Article PubMed Google Scholar -
Johnson LA, June CH. Driving gene-engineered T cell immunotherapy of cancer. Cell research. 2017;27(1):38-58.
.
View Article PubMed Google Scholar -
Schendel DJ, Frankenberger B. Limitations for TCR gene therapy by MHC-restricted fratricide and TCR-mediated hematopoietic stem cell toxicity. Oncoimmunology. 2013;2(1):e22410.
.
View Article PubMed Google Scholar -
Barrett DM, Grupp SA, June CH. Chimeric antigen receptor-and TCR-modified T cells enter main street and wall street. The Journal of Immunology. 2015;195(3):755-61.
.
View Article PubMed Google Scholar -
González-Galarza FF, Takeshita LY, Santos EJ, Kempson F, Maia MHT, Silva ALSd, et al. Allele frequency net 2015 update: new features for HLA epitopes, KIR and disease and HLA adverse drug reaction associations. Nucleic acids research. 2015;43(D1):D784-D8.
.
View Article PubMed Google Scholar -
Okamoto S, Mineno J, Ikeda H, Fujiwara H, Yasukawa M, Shiku H, et al. Improved expression and reactivity of transduced tumor-specific TCRs in human lymphocytes by specific silencing of endogenous TCR. Cancer research. 2009;69(23):9003-11.
.
View Article PubMed Google Scholar -
Provasi E, Genovese P, Lombardo A, Magnani Z, Liu P-Q, Reik A, et al. Editing T cell specificity towards leukemia by zinc finger nucleases and lentiviral gene transfer. Nat Med. 2012;18(5):807.
.
View Article PubMed Google Scholar -
Fontaine P, Roy-Proulx G, Knafo L, Baron C, Roy D-C, Perreault C. Adoptive transfer of minor histocompatibility antigen-specific T lymphocytes eradicates leukemia cells without causing graft-versus-host disease. Nat Med. 2001;7(7):789-94.
.
View Article PubMed Google Scholar -
Granados D, Rodenbrock A, Laverdure J, Cote C, Caron-Lizotte O, Carli C, et al. Proteogenomic-based discovery of minor histocompatibility antigens with suitable features for immunotherapy of hematologic cancers. Leukemia. 2016;30(6):1344-54.
.
View Article PubMed Google Scholar -
Montel-Hagen A, Seet CS, Li S, Chick B, Zhu Y, Chang P, et al. Organoid-induced differentiation of conventional T cells from human pluripotent stem cells. Cell stem cell. 2019;24(3):376-89. e8.
.
View Article PubMed Google Scholar -
Bonte S, De Munter S, Goetgeluk G, Ingels J, Pille M, Billiet L, et al. T-cells with a single tumor antigen-specific T-cell receptor can be generated in vitro from clinically relevant stem cell sources. Oncoimmunology. 2020;9(1):1727078.
.
View Article PubMed Google Scholar -
Kuwana Y, Asakura Y, Utsunomiya N, Nakanishi M, Arata Y, Itoh S, et al. Expression of chimeric receptor composed of immunoglobulin-derived V resions and T-cell receptor-derived C regions. Biochemical and biophysical research communications. 1987;149(3):960-8.
.
View Article Google Scholar -
Gross G, Waks T, Eshhar Z. Expression of immunoglobulin-T-cell receptor chimeric molecules as functional receptors with antibody-type specificity. Proceedings of the National Academy of Sciences. 1989;86(24):10024-8.
.
View Article PubMed Google Scholar -
Van Der Stegen SJ, Hamieh M, Sadelain M. The pharmacology of second-generation chimeric antigen receptors. Nature reviews Drug discovery. 2015;14(7):499-509.
.
View Article PubMed Google Scholar -
Zheng P-P, Kros JM, Li J. Approved CAR T cell therapies: ice bucket challenges on glaring safety risks and long-term impacts. Drug discovery today. 2018;23(6):1175-82.
.
View Article PubMed Google Scholar -
Tang X-Y, Sun Y, Zhang A, Hu G-L, Cao W, Wang D-H, et al. Third-generation CD28/4-1BB chimeric antigen receptor T cells for chemotherapy relapsed or refractory acute lymphoblastic leukaemia: a non-randomised, open-label phase I trial protocol. BMJ open. 2016;6(12).
.
View Article PubMed Google Scholar -
Ramos CA, Rouce R, Robertson CS, Reyna A, Narala N, Vyas G, et al. In vivo fate and activity of second-versus third-generation CD19-specific CAR-T cells in B cell non-Hodgkin's lymphomas. Molecular Therapy. 2018;26(12):2727-37.
.
View Article PubMed Google Scholar -
Chmielewski M, Abken H. TRUCKs: the fourth generation of CARs. Expert opinion on biological therapy. 2015;15(8):1145-54.
.
View Article PubMed Google Scholar -
Kueberuwa G, Kalaitsidou M, Cheadle E, Hawkins RE, Gilham DE. CD19 CAR T cells expressing IL-12 eradicate lymphoma in fully lymphoreplete mice through induction of host immunity. Molecular Therapy-Oncolytics. 2018;8:41-51.
.
View Article PubMed Google Scholar -
Eapen M, Rocha V, Sanz G, Scaradavou A, Zhang M-J, Arcese W, et al. Effect of graft source on unrelated donor haemopoietic stem-cell transplantation in adults with acute leukaemia: a retrospective analysis. The lancet oncology. 2010;11(7):653-60.
.
View Article Google Scholar -
Kwoczek J, Riese SB, Tischer S, Bak S, Lahrberg J, Oelke M, et al. Cord blood-derived T cells allow the generation of a more naïve tumor‐reactive cytotoxic T‐cell phenotype. Transfusion. 2018;58(1):88-99.
.
View Article PubMed Google Scholar -
Kadereit S, Mohammad SF, Miller RE, Woods KD, Listrom CD, McKinnon K, et al. Reduced NFAT1 protein expression in human umbilical cord blood T lymphocytes. Blood, The Journal of the American Society of Hematology. 1999;94(9):3101-7.
.
View Article PubMed Google Scholar -
Themeli M, Kloss CC, Ciriello G, Fedorov VD, Perna F, Gonen M, et al. Generation of tumor-targeted human T lymphocytes from induced pluripotent stem cells for cancer therapy. Nature biotechnology. 2013;31(10):928.
.
View Article PubMed Google Scholar -
Prentice H. OKT3 incubation of donor marrow for prophylaxis of acute graft-vs.-host disease (GvHD) in allogeneic bone marrow transplantation. Journal of clinical immunology. 1982;2(3 Suppl):148S-53S.
.
-
Champlin RE, Passweg JR, Zhang M-J, Rowlings PA, Pelz CJ, Atkinson KA, et al. T-cell depletion of bone marrow transplants for leukemia from donors other than HLA-identical siblings: advantage of T-cell antibodies with narrow specificities. Blood, The Journal of the American Society of Hematology. 2000;95(12):3996-4003.
.
-
Brudno JN, Somerville RP, Shi V, Rose JJ, Halverson DC, Fowler DH, et al. Allogeneic T cells that express an anti-CD19 chimeric antigen receptor induce remissions of B-cell malignancies that progress after allogeneic hematopoietic stem-cell transplantation without causing graft-versus-host disease. Journal of clinical oncology. 2016;34(10):1112.
.
View Article PubMed Google Scholar -
Münz C. Redirecting T Cells against Epstein-Barr Virus Infection and Associated Oncogenesis. Cells. 2020;9(6):1400.
.
View Article PubMed Google Scholar -
Chu J, Deng Y, Benson DM, He S, Hughes T, Zhang J, et al. CS1-specific chimeric antigen receptor (CAR)-engineered natural killer cells enhance in vitro and in vivo antitumor activity against human multiple myeloma. Leukemia. 2014;28(4):917-27.
.
View Article PubMed Google Scholar -
Liu E, Tong Y, Dotti G, Shaim H, Savoldo B, Mukherjee M, et al. Cord blood NK cells engineered to express IL-15 and a CD19-targeted CAR show long-term persistence and potent antitumor activity. Leukemia. 2018;32(2):520-31.
.
View Article PubMed Google Scholar -
Chaidos A, Patterson S, Szydlo R, Chaudhry MS, Dazzi F, Kanfer E, et al. Graft invariant natural killer T-cell dose predicts risk of acute graft-versus-host disease in allogeneic hematopoietic stem cell transplantation. Blood, The Journal of the American Society of Hematology. 2012;119(21):5030-6.
.
View Article PubMed Google Scholar -
Rotolo A, Caputo VS, Holubova M, Baxan N, Dubois O, Chaudhry MS, et al. Enhanced anti-lymphoma activity of CAR19-iNKT cells underpinned by dual CD19 and CD1d targeting. Cancer Cell. 2018;34(4):596-610. e11.
.
View Article PubMed Google Scholar -
Capsomidis A, Benthall G, Van Acker HH, Fisher J, Kramer AM, Abeln Z, et al. Chimeric antigen receptor-engineered human gamma delta T cells: enhanced cytotoxicity with retention of cross presentation. Molecular Therapy. 2018;26(2):354-65.
.
View Article PubMed Google Scholar -
Qasim W, Amrolia PJ, Samarasinghe S, Ghorashian S, Zhan H, Stafford S, et al. First clinical application of Talen engineered universal CAR19 T cells in B-ALL. American Society of Hematology Washington, DC; 2015.
.
View Article Google Scholar -
Torikai H, Reik A, Liu P-Q, Zhou Y, Zhang L, Maiti S, et al. A foundation for universal T-cell based immunotherapy: T cells engineered to express a CD19-specific chimeric-antigen-receptor and eliminate expression of endogenous TCR. Blood. 2012;119(24):5697-705.
.
View Article PubMed Google Scholar -
Eyquem J, Mansilla-Soto J, Giavridis T, van der Stegen SJ, Hamieh M, Cunanan KM, et al. Targeting a CAR to the TRAC locus with CRISPR/Cas9 enhances tumour rejection. Nature. 2017;543(7643):113-7.
.
View Article PubMed Google Scholar -
Qasim W, Zhan H, Samarasinghe S, Adams S, Amrolia P, Stafford S, et al. Molecular remission of infant B-ALL after infusion of universal TALEN gene-edited CAR T cells. Science translational medicine. 2017;9(374).
.
View Article PubMed Google Scholar -
Jain N, Graham C, Konopleva M. UCART19, an allogeneic anti-CD19 CAR T-cell product, in high risk adult patients with CD19+ relapsed/refractory B-cell acute lymphoblastic leukemia: preliminary results of phase I CALM study (Poster). EHA https://learningcenter ehaweb org/eha/2018/stockholm/214674/reuben benjamin ucart19 an allogeneic anti-cd19 car t-cell product in high html. 2018..
.
Comments
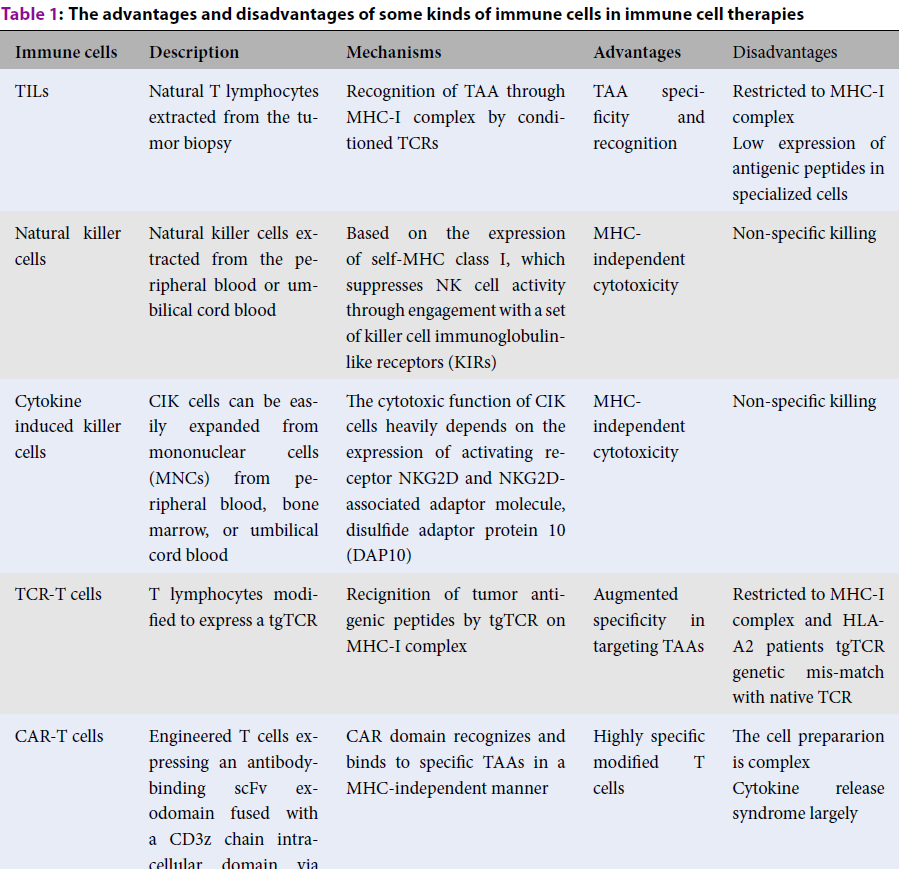
Downloads
Article Details
Volume & Issue : Vol 7 No 12 (2020)
Page No.: 4170-4189
Published on: 2020-12-31
Citations
Copyrights & License
This work is licensed under a Creative Commons Attribution 4.0 International License.
Search Panel
Pubmed
Google Scholar
Pubmed
Google Scholar
Pubmed
Google Scholar
Pubmed
Google Scholar
Pubmed
Google Scholar
Pubmed
Search for this article in:
Google Scholar
Researchgate
- HTML viewed - 8605 times
- Download PDF downloaded - 1889 times
- View Article downloaded - 0 times
