

Copyrights: Phuong Thu Vu Hoang, Minh Xuan Ngo, Quang Minh Le Tran, Thanh Vinh Nguyen, Thanh Vu Nguyen, Anh Thu Ha Nguyen, Thu Hang Do Thi, Dieu Hien Huynh Thi, Chung Thuy Tran Phan, 2023. License: This work is licensed under a Creative Commons Attribution 4.0 International License.
Abstract
Introduction: Non-syndromic hearing loss (NSHL) in children, which has numerous causes, can impede or even postpone the acquisition of spoken language. In Viet Nam, screening programs and genetic testing for NSHL are rarely applied. In this study, 31 pediatric patients had their medical histories collected alongside sequencing results for the GJB2 and TECTA genes to determine the prevalence of these mutations in the community and their associations with potential risk factors.
Methods: Information and blood samples were collected from 31 severe-to-profound pediatric NSHL patients. DNA was extracted, amplified by polymerase chain reaction (PCR), and directly sequenced for the detection of GJB2 (connexin 26) and TECTA mutations.
Results: No TECTA gene mutations were detected. GJB2 mutations were identified in eight patients (25.8%), with three (9.7%) cases of heterozygous c.109G>A (V37I), four (12.9%) cases of homozygous c.109G>A (V37I), and one (3.2%) case of heterozygous c.299-300delAT. There were no significant associations between having mutated GJB2 genes and living in urban areas, having a family history of prelingual deafness, or having an abnormal obstetric history (p > 0.05, Fisher's exact tests).
Conclusion: Our study addresses the high prevalence of GJB2 mutations as causative factors in hearing loss in diagnosed patients at the Otorhino-laryngology Hospital in Ho Chi Minh City, Viet Nam. Further studies are required to obtain a better understanding of the genetic spectrum of NSHL and to articulate its relationship with various risk factors.
Introduction
Hearing loss is a condition of partially or totally losing the ability to hear. Nearly 500 million people around the world have hearing loss1. Among children, 1.1 per 1000 newborns are diagnosed with severe or even profound bilateral hearing loss2. This condition of deficiency in children impedes or even postpones the acquisition of spoken language3. Additionally, hearing impairment can also negatively affect a child’s learning ability and educational achievements4.
There are numerous causes of hearing loss, but more than 50% are related to genes and gene mutations5. Many genes are involved in this process, and research has shown that mutations in the GJB2 gene are the most common in non-syndromic hearing loss (NSHL)6, 7. GJB2 has mutations that replace the protein building block glycine with serine at protein position 59 (Gly59Ser or G59S) or replace asparagine with lysine at protein position 54. These replacements disrupt the function of connexin 26, which can lead to skin growth abnormalities and hearing impairment by disturbing the conversion of sound waves to nerve impulses. Meanwhile, the TECTA gene has a role in synthesizing the alpha-tectorin protein, which is part of the tectorial membrane in the inner ear. TECTA mutations, which have recently been discovered, have been found to be the fourth most common factor in NSHL6, 8. Research in the Vietnamese population has shown that the molecular diagnostic yield rate in severe-to-profound hearing loss patients is 31.7%, with the GJB2 mutation being the second most prevalent (6.9%) after the MYO15A gene (7.2%)9.
Early diagnosis of NSHL and interventions, such as early hearing aids or early cochlear implants, can effectively improve speech and language skills10. However, in developing countries, including Viet Nam, screening programs for congenital deafness are not widely implemented, especially since genetic diagnostic screening is expensive and rarely applied. Therefore, it is important to develop a better understanding of the prevalence of genetic mutations in hearing loss in children while also being able to examine its associations with potential risk factors. This will provide a firm foundation for the establishment of an adequate screening program. In this study, 49 pediatric patients had their medical histories collected and analyzed alongside sequencing results of the GJB2 and TECTA genes to determine the prevalence of these mutations in the community, as well as their association with certain risk factors, including living environment (urban or rural), obstetric history, blood type, and family history of hearing loss.
Methods
Data collection
A cross-sectional study using a random sampling technique was conducted at the Otorhino-laryngology Hospital in Ho Chi Minh City, Viet Nam from July 2018 to October 2022. Pediatric patients with a medical history of hearing impairment were selected for further investigation. The patients or their parents were interviewed for medical history and family history of hearing loss before clinical examinations were conducted, which included the tympanometry test, acoustic reflex (AR) test, otoacoustic emissions (OAE) test, and auditory brainstem response (ABR) test. Patients who were diagnosed with profound bilateral hearing loss (110 dB or more and absent OAE) had their blood samples collected for polymerase chain reaction (PCR) and direct DNA sequencing for GJB2 (connexin 26) and TECTA mutations. The results were collected and analyzed using SnapGene software.
Statistical analysis
Data were organized in a spreadsheet using Microsoft Excel and analyzed using IBM SPSS Statistics, Version 20.0 (IBM Corp., Armonk, NY, USA). Percentages were reported for qualitative variables, while mean (and standard deviation) or median (and interquartile range) were used for quantitative variables (depending on data distribution, confirmed by the Shapiro–Wilk normality test). Prevalence ratios (with 95% confidence intervals) and the chi-square test or Fisher’s exact test (if applicable) were used to determine significant differences in risk factors between patients with and without gene mutations. A p-value < 0.05 was considered statistically significant.
DNA isolation
For each subject, one venous blood sample (2 – 3 mL) was collected in an EDTA tube. Genomic DNA was extracted from peripheral blood lymphocytes using the E.Z.N.A. DNA mini Blood Kit (Omega Bio-Tek Inc., Norcross, GA, USA), according to the manufacturer’s protocol. Concentrations of extracted DNA were stored at −20°C until use.
Pyrosequencing
Pyrosequencing was performed on a 3500 Genetic Analyzer (Applied Biosystems/Thermo Fisher Scientific, Waltham, MA, USA). The primers used for PCR and pyrosequencing (IDT Integrated DNA Technologies, Coralville, IA, USA), as well as the sizes of the corresponding amplicons, are presented in Table 1. The PCR reaction mixture was as follows: 10 µL Qiagen Master Mix (Hilden, Germany), 3 µL nuclease-free water, 1 µL of each primer, and 5 µL (~150 – 200 ng) of genomic DNA in a final volume of 50 µL. The cycling conditions were 95°C for 15 minutes, followed by 40 cycles at 95°C for 15 seconds, 52°C for 45 seconds, and 72°C for 60 seconds, with a final extension step at 72°C for 5 minutes.
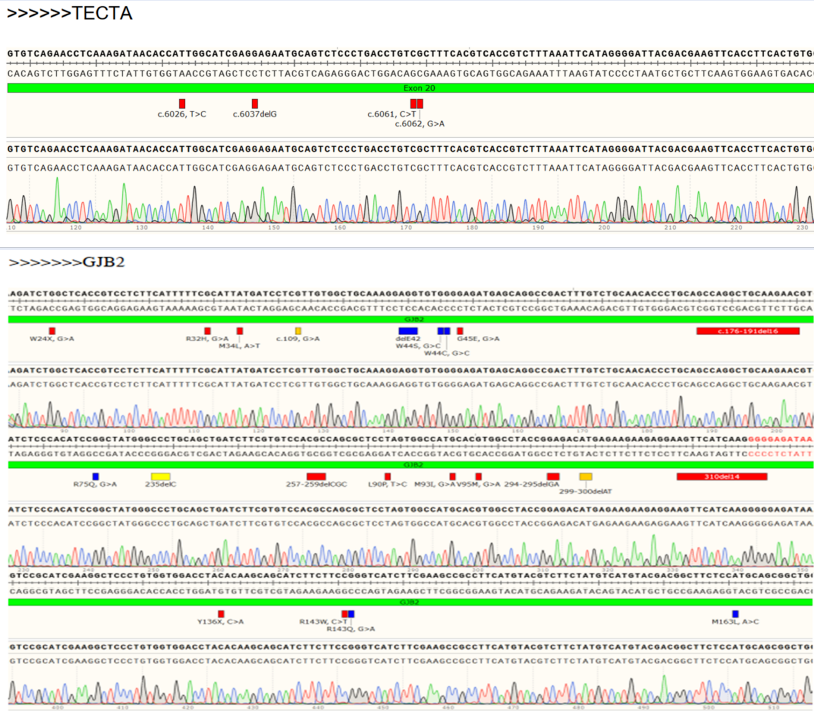
For each target gene, a sequencing primer was further hybridized to the PCR product and incubated with reagents containing enzymes and substrates. One dNTP was added to the reaction mixture at a time and incorporated into the DNA strand if it was complementary to the base in the template strand. The incorporation resulted in a release of pyrophosphate that was converted to ATP, which in turn drove the conversion of the substrate luciferin to oxyluciferin, thereby generating light corresponding to a peak (Figure 1).
| Target gene | Description | Location | Primers | Forward and reverse primers (5’–3’) used in polymerase chain reaction | Target size (bp) |
|---|---|---|---|---|---|
| GJB2 | Gap junction beta-2 protein | 13q12.11 | GJB2-F | GCATTCGTCTTTTCCAGAGCAAACCGCCC | 814 bp |
| GJB2-R | GGGAAATGCTAGCGACTGAGCCTTGACAGC | ||||
| TECTA | Alpha-tectorin protein | 11q23.3 | TECTA-F | AGTCCAGGGGTCACTTTCAAATGTAAAGGCA | 523 bp |
| TECTA-R | GCGTTAAGCCAATGACTGGTTGACAATTC |
| Risk factors | Number of cases (N = 49) | With GJB2 mutation (N = 12) | PR (95% CI) | p-value |
|---|---|---|---|---|
| Urban environment | 30 (61.2%) | 9 (75.0%) | 1.900 (0.588–6.143) | 0.323* |
| Family history of prelingual hearing loss | 3 (6.1%) | 1 (8.3%) | 1.394 (0.259–7.489) | 1.000* |
| Abnormal obstetric history | 21 (42.9%) | 6 (50.0%) | 1.333 (0.500–3.553) | 0.565 |
| Rash | 9 (18.4%) | 1 (8.3%) | 0.404 (0.060–2.743) | 0.420* |
| Fever | 7 (14.3%) | 1 (8.3%) | 0.545 (0.083–3.590) | 0.665* |
| First trimester | 3 (6.1%) | 0 (0.0%) | 0.566* | |
| Second trimester | 3 (6.1%) | 0 (0.0%) | 0.566* | |
| Third trimester | 1 (2.0%) | 1 (8.3%) | 4.364 (2.597–7.331) | 0.245* |
| Preterm birth | 8 (16.3%) | 4 (33.3%) | 2.563 (1.010–6.501) | 0.088* |
| Cesarean section | 9 (18.4%) | 2 (16.7%) | 0.889 (0.234–3.378) | 1.000* |
| Blood type (Two cases were missing, N = 47, 11 cases with GJB2 mutation) | ||||
| A+ | 9 (19.1%) | 2 (18.2%) | 0.938 (0.243–3.615) | 1.000* |
| B+ | 20 (42.6%) | 5 (45.5%) | 1.125 (0.399–3.172) | 1.000* |
| AB+ | 2 (4.3%) | 1 (9.1%) | 2.250 (0.507–9.981) | 0.417* |
| O+ | 16 (34.0%) | 3 (27.3%) | 0.727 (0.223–2.369) | 0.725* |


Results
A total of 49 pediatric patients with severe-to-profound hearing loss were included in this study. There were 23 (46.9%) males and 26 (53.1%) females. The median age was 5 (3–11) years old (not normally distributed, p < 0.001, Shapiro–Wilk test), and the age range was 1–18 years old. Most patients were diagnosed with hearing loss at the age of 2 years or younger, with 30.6% being diagnosed before 1 year of age; 24.5% and 32.7% were diagnosed at 1 and 2 years old, respectively. The oldest age at the time of diagnosis was 12 years. The median age of cochlear implantation was 6 (3–11) years, with a range from 1 to 18 years (not normally distributed, p = 0.002, Shapiro–Wilk test) (Table 2).
Regarding risk factors, there were 30 (61.2%) patients from urban regions. Three (6.1%) cases had a family history of prelingual hearing loss, and 21 (42.9%) cases had an abnormal obstetric history. The abnormal obstetric events of the mothers were recorded as follows: Cesarean birth and having rashes during pregnancy accounted for 18.4% each, followed by 16.3% who had preterm birth, and 14.3% who had a fever during pregnancy. Blood type information was also collected. Blood types B and O were the two most prevalent (42.6% and 34.0%, respectively), followed by 19.1% for type A, and 4.3% for type AB. All cases were Rh-positive. Despite having the lowest prevalence, the rate of carrying a GJB2 mutation in children with AB-positive blood was notably high (50%). As for mutational screening/sequencing results, no TECTA gene mutations were detected. However, GJB2 mutations were identified in 12 patients, accounting for 24.5% of all cases. There were four (8.2%) cases of heterozygous c.109G>A (V37I), four (8.2%) cases of homozygous c.109G>A (V37I), one (2.0%) case of heterozygous c.299-300delAT, one (2.0%) case of heterozygous c.341A>G, one (2.0%) case of heterozygous c.235DelC combined with heterozygous c.364DelA, and one case (2.0%) of heterozygous c.109G>A combined with heterozygous c.299-300DelAT. The number of cases with GJB2 mutations separated by gender are demonstrated in Figure 2. There were 7 boys with GJB2 mutations, with 3 cases of heterozygous c.314A>G, heterozygous c.109G>A combined with heterozygous c.299-300DelAT, and heterozygous c.299-300delAT were only identified in male patients. In comparison, 5 girls were diagnosed with GJB2 mutations, and one case of heterozygous c.235DelC combined with heterozygous c.364DelA was diagnosed in female patient only.When analyzed with risk factor data, preterm birth may potentially be associated with carrying a GJB2 mutation in children with profound bilateral hearing loss, with a prevalence ratio of 2.563 (95% CI: 1.010–6.501), even though p = 0.088 (p < 0.05). No significant association was found between having GJB2 mutated genes and other factors, such as living in urban areas, having a family history of prelingual deafness, having an abnormal obstetric history, or having any specific blood type (p > 0.05, chi-square test or Fisher’s exact test) (Table 2).
Discussion
Hereditary hearing loss is one of the most common congenital disorders, with approximately 3 out of 1,000 newborns having reduced bilateral hearing sensitivity at the time of screening. In developing countries like Vietnam, congenital hearing loss can be affected by environmental and hereditary factors, with the latter involved in nearly 50–60% of cases11. Establishing a genetic basis plays a crucial role in the diagnosis, intervention, and treatment of congenital NSHL. However, this process can be difficult since more than 300 genes are considered to be related to NSHL due to the complex structure of the hearing system12. Therefore, knowing which genes to target and which children to test is crucial to delivering an effective and achievable screening program.
Only pediatric patients were included in our study, with a median age of 5 (3–11) years old. Interestingly, the age at time of hearing loss diagnosis was most often 2 years or even younger, while the median age of intervention was 6 (3–11) years, suggesting that hearing loss treatment was delayed, which could negatively affect the acquisition of speech and learning abilities. Treatment costs may be the primary cause of delays in a developing country like Viet Nam. Further research is required to draw a conclusion on this matter and to establish a basis for the development of medical policies.
Regarding the genetic etiology of hearing loss, GJB2 mutations were identified in 24.5% of our cases. Our result was remarkably higher when compared to a prevalence of just 6.9% from a study in Children 1 Hospital—Ho Chi Minh City in Viet Nam in 2016; notably, that study included all severities of hearing loss9. A study conducted at the University of Iowa in 2016 showed a very similar result to our finding of a rate of 21.6%6. Research from China published in 2002 found a GJB2 mutation rate of approximately 20%13. A proportion of 18.17% of deaf Iranian patients had GJB2 mutations7. These results suggest that GJB2 mutations were still the most frequent causative factor in genetic-related deafness in children.
In our study, in detail, GJB2 mutations included four (8.2%) cases identified as heterozygous c.109G>A (V37I), four (8.2%) cases were homozygous c.109G>A (V37I), and one case (2.0%) was heterozygous c.109G>A combined with heterozygous c.299-300DelAT (Figure 2). Overall, this result is generally higher compared to results from a study at Chinese PLA General Hospital, which found that 8.13% of patients had GJB2 p.V37I variations, with heterozygous p.V37I in 5.38% of cases, homozygous p.V37I in 1.48% of cases, and compound p.V37I plus other GJB2 mutations in 1.27% of cases14. Research from Children 1 Hospital—Ho Chi Minh City, Viet Nam showed a prevalence of 4.6% for the homozygous p.V37I variation of GJB2 and one (1.1%) compound heterozygous V37I/235delC case9. Additionally, one (2.0%) case of heterozygous c.299-300delAT, one (2.0%) case of heterozygous c.341A>G, and one (2.0%) case of compound mutation (heterozygous c.235DelC combined with heterozygous c.364DelA) were detected in our study. The detection rate of the c.299-300delAT mutation was also low in other studies. For example, only one (2.2%) case of homozygous c.299-300delAT was detected in a study of the Kurdish population in Iran15. A study conducted in China of 35 severe-to-profound deaf patients detected only one case of c.299-300delAT16. For heterozygous c.341A>G, we identified one case of a single mutation. However, this type is often reported in the form of compound mutations. A study at Children 1 Hospital—Ho Chi Minh City, Viet Nam conducted on 96 non-syndromic congenital hearing loss patients identified 15 (16.67%) cases of compound mutations, which included heterozygous c.341A>G17. A study of 155 hearing impaired patients (not restricted to children) in China found 37 cases of heterozygous c.341A>G, with only one case having no combined mutations18. This may imply the rarity of heterozygous c.341A>G being an independent factor in hearing loss screening. Interestingly, a case of heterozygous c.235DelC plus heterozygous c.364DelA might be a novel finding since heterozygous c.235DelC is often referred to as a singular mutation on an allele17, 18; until now, no cases of NSHL children with heterozygous c.364DelA mutation have been reported.
The TECTA mutation was not detected in our study. According to other studies, its prevalence can be very low, such as 5.2% in the University of Iowa study6, or as low as 3.2% in research from Japan19. While the TECTA mutation is also one of the more frequent mutations6, a larger sample size might be necessary to identify such rare cases.
Researching risk factors for genetic hearing loss is an interesting topic, but to our knowledge, very few reports have been published on this matter. Due to limited sample size, our data might act as a pilot study for risk factors. Our study showed no significant associations between having GJB2 mutations and living in an urban environment, having a family history of prelingual deafness, or having an abnormal obstetric history (p > 0.05, Fisher’s exact test). However, preterm birth may potentially be a risk factor for carrying a GJB2 mutation, as data show a prevalence ratio of 2.563 (95% CI: 1.010–6.501) and a low p-value of 0.088. In comparison, a study of severe-to-profound deaf patients in the Romanian population, which reported a GJB2 carrier rate of 44.12%, showed that prematurity and living in an urban environment can be risk factors for a genetic etiology of hearing loss20.
Our study found that the rate of NSHL children with B-positive blood type was the highest (42.6%) compared to O-positive blood type in the general Vietnamese population. The rate of children with AB-positive blood carrying a GJB2 mutation was quite high (50%), although the sample size was very small (only two cases), and the finding was not statistically significant (p = 0.417). Due to our study design and limited sample size, the association between B-positive blood and NSHL, as well as between AB-positive blood and being a mutated GJB2 carrier, cannot be further tested to either confirm or rule out these possible associations, so a case-control study with an adequate sample size is required to elucidate this matter.
Our study has several weaknesses that need to be addressed. First, the sample size is very small, which made it difficult to detect TECTA mutations and to draw any definitive conclusions about risk factors. Second, due to limited resources, our study did not include a wide range of genetic mutations but focused only on the GJB2 and TECTA genes, so the full picture of the genetic etiology of pediatric hearing loss might not be clearly apparent. Nevertheless, this is one of a few studies that establish a foundation for genetic research on NSHL in Ho Chi Minh City, Viet Nam. In particular, it points out that there has been delayed treatment in diagnosed children and provides a primitive look into the relationship between hearing loss in children and possible risk factors.
Conclusions
Hearing loss at an early age exposes patients to several drawbacks if they are not diagnosed and treated appropriately and in a timely manner. Our study once again addresses the high prevalence of GJB2 mutations as a causative factor in hearing loss, identifies one case of NSHL with a novel mutation (heterozygous c.235DelC + heterozygous c.364DelA), and highlights the delay in treatment in diagnosed patients at the Otorhino-laryngology Hospital in Ho Chi Minh City, Viet Nam. Further studies are required to develop a better understanding of the genetic spectrum of NSHL and to articulate its relationship with several risk factors, ultimately establishing a foundation for the development of prophylaxis and treatment programs.
Abbreviations
ABR: Auditory Brainstem Response, AR: Acoustic Reflex, GJB2: Gap junction protein beta 2, NSHL: Non-Syndromic Hearing Loss, OAE: Otoacoustic Emission, PCR: Polymerase Chain Reaction, TECTA: Tectorin alpha
Acknowledgments
We thank all the patients and the families for their participation in our study.
Author’s contributions
Phuong Thu Vu Hoang: analysis, interpretation of data for the work, drafting the work; final approval of the version to be published; Minh Xuan Ngo, Quang Minh Le Tran, Thanh Vinh Nguyen: Substantial contributions to the conception or design of the work; Hong Giang Do, Thanh Vu Nguyen, Thu Hang Do Thi, Dieu Hien Huynh Thi: Agreement to be accountable for all aspects of the work in ensuring that questions related to the accuracy or integrity of any part of the work are appropriately investigated and resolved; Anh Thu Ha Nguyen: Drafting the work
Funding
This research is funded by Vietnam National University HoChiMinh City (VNU-HCM) under grant number GENE2020-44-01.
Availability of data and materials
Data and materials used and/or analyzed during the current study are available from the corresponding author on reasonable request.
Ethics approval and consent to participate
This study was conducted in accordance with the amended Declaration of Helsinki. The institutional review board approved the study (number of ethical approval 53/HĐĐĐ- TĐHYKPNT, 20 June 2019), and all participants provided written informed consent.
Consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing interests.
References
-
Wilson
B.S.,
Tucci
D.L.,
Merson
M.H.,
O'Donoghue
G.M.,
Global hearing health care: new findings and perspectives. Lancet.
2017;
390
(10111)
:
2503-15
.
View Article PubMed Google Scholar -
Butcher
E.,
Dezateux
C.,
Cortina-Borja
M.,
Knowles
R.L.,
Prevalence of permanent childhood hearing loss detected at the universal newborn hearing screen: systematic review and meta-analysis. PLoS One.
2019;
14
(7)
:
e0219600
.
View Article PubMed Google Scholar -
Tomblin
J.B.,
Harrison
M.,
Ambrose
S.E.,
Walker
E.A.,
Oleson
J.J.,
Moeller
M.P.,
Language Outcomes in Young Children with Mild to Severe Hearing Loss. Ear and Hearing.
2015;
36
(0 1)
:
76-91
.
View Article PubMed Google Scholar -
Marschark
M.,
Shaver
D.M.,
Nagle
K.M.,
Newman
L.A.,
Predicting the Academic Achievement of Deaf and Hard-of-Hearing Students From Individual, Household, Communication, and Educational Factors. Exceptional Children.
2015;
81
(3)
:
350-69
.
View Article PubMed Google Scholar -
Khela
H.,
Kenna
M.A.,
Genetics of pediatric hearing loss: A functional perspective. Laryngoscope Investigative Otolaryngology.
2020;
5
(3)
:
511-9
.
View Article PubMed Google Scholar -
Sloan-Heggen
C.M.,
Bierer
A.O.,
Shearer
A.E.,
Kolbe
D.L.,
Nishimura
C.J.,
Frees
K.L.,
Comprehensive genetic testing in the clinical evaluation of 1119 patients with hearing loss. Human Genetics.
2016;
135
(4)
:
441-50
.
View Article PubMed Google Scholar -
Mahdieh
N.,
Rabbani
B.,
Wiley
S.,
Akbari
M.T.,
Zeinali
S.,
Genetic causes of nonsyndromic hearing loss in Iran in comparison with other populations. Journal of Human Genetics.
2010;
55
(10)
:
639-48
.
View Article PubMed Google Scholar -
Dodson
K.M.,
Georgolios
A.,
Barr
N.,
Nguyen
B.,
Sismanis
A.,
Arnos
K.S.,
Etiology of unilateral hearing loss in a national hereditary deafness repository. American Journal of Otolaryngology.
2012;
33
(5)
:
590-4
.
View Article PubMed Google Scholar -
Han
J.J.,
Nguyen
P.D.,
Oh
D.Y.,
Han
J.H.,
Kim
A.R.,
Kim
M.Y.,
Elucidation of the unique mutation spectrum of severe hearing loss in a Vietnamese pediatric population. Scientific Reports.
2019;
9
(1)
:
1604
.
View Article PubMed Google Scholar -
Lieu
J.E.,
Kenna
M.,
Anne
S.,
Davidson
L.,
Hearing Loss in Children: A Review. Journal of the American Medical Association.
2020;
324
(21)
:
2195-205
.
View Article PubMed Google Scholar -
Morton
C.C.,
Nance
W.E.,
Newborn hearing screening - silent revolution. The New England Journal of Medicine.
2006;
354
(20)
:
2151-64
.
View Article PubMed Google Scholar -
Lin
X.,
Tang
W.,
Ahmad
S.,
Lu
J.,
Colby
C.C.,
Zhu
J.,
Applications of targeted gene capture and next-generation sequencing technologies in studies of human deafness and other genetic disabilities. Hearing Research.
2012;
288
(1-2)
:
67-76
.
View Article PubMed Google Scholar -
Liu
Y.,
Ke
X.,
Qi
Y.,
Li
W.,
Zhu
P.,
Connexin26 gene ( GJB2): prevalence of mutations in the Chinese population. Journal of Human Genetics.
2002;
47
(12)
:
688-90
.
View Article PubMed Google Scholar -
Huang
S.,
Huang
B.,
Wang
G.,
Yuan
Y.,
Dai
P.,
The Relationship between the p.V37I Mutation in GJB2 and Hearing Phenotypes in Chinese Individuals. PLoS One.
2015;
10
(6)
:
e0129662
.
View Article PubMed Google Scholar -
Azadegan-Dehkordi
F.,
Bahrami
T.,
Shirzad
M.,
Karbasi
G.,
Yazdanpanahi
N.,
Farrokhi
E.,
Mutations in GJB2 as Major Causes of Autosomal Recessive Non-Syndromic Hearing Loss: First Report of c.299-300delAT Mutation in Kurdish Population of Iran. Journal of Audiology & Otology.
2019;
23
(1)
:
20-6
.
View Article PubMed Google Scholar -
Chen
G.,
Fu
S.,
Dong
J.,
Chen
P.,
Low frequency of GJB2 mutations in thirty-five students with hearing loss in Chinese consanguineous families. International Journal of Pediatric Otorhinolaryngology.
2011;
75
(12)
:
1535-7
.
View Article PubMed Google Scholar -
Dinh
N.,
Nguyen
D.H.,
Huyen Lam
T.,
Tho Nguyen
K.,
Dinh Nguyen
C.,
Nguyen
H.,
Mutational Analysis of GJB2 Gene in Non-Syndromic Hearing Loss from Patients at Children's Hospital 1- Ho Chi Minh City, Vietnam. SM Otolaryngology..
2017;
1
(2)
:
1-6
.
View Article Google Scholar -
Jiang
Y.,
Huang
S.,
Deng
T.,
Wu
L.,
Chen
J.,
Kang
D.,
Mutation Spectrum of Common Deafness-Causing Genes in Patients with Non-Syndromic Deafness in the Xiamen Area, China. PLoS One.
2015;
10
(8)
:
e0135088
.
View Article PubMed Google Scholar -
Yasukawa
R.,
Moteki
H.,
Nishio
S.Y.,
Ishikawa
K.,
Abe
S.,
Honkura
Y.,
The Prevalence and Clinical Characteristics of TECTA-Associated Autosomal Dominant Hearing Loss. Genes.
2019;
10
(10)
:
744
.
View Article PubMed Google Scholar -
Neagu
A.,
Mocanu
A.I.,
Bonciu
A.,
Coad\ua
G.,
Mocanu
H.,
Prevalence of GJB2 gene mutations correlated to presence of clinical and environmental risk factors in the etiology of congenital sensorineural hearing loss of the Romanian population. Experimental and Therapeutic Medicine.
2021;
21
(6)
:
612
.
View Article PubMed Google Scholar
Comments
Article Details
Volume & Issue : Vol 10 No 1 (2023)
Page No.: 5523-5529
Published on: 2023-01-31
Citations
Copyrights & License
This work is licensed under a Creative Commons Attribution 4.0 International License.
Search Panel
- HTML viewed - 4351 times
- PDF downloaded - 1242 times
- XML downloaded - 0 times
