

Copyrights: Gloria Agrimonti, Giorgia Spaggiari, Soara Menabò, Silvia Vezzani, Elisa Magnani, Andrea Frasoldati, Antonio R.M. Granata, Daniele Santi, 2024. License: This work is licensed under a Creative Commons Attribution 4.0 International License.
Abstract
Background: Classical congenital adrenal hyperplasia is a genetic disorder characterized by defects in the steroidogenesis cascade, mainly due to 21-hydroxylase enzyme deficiency. The phenotype can vary from the most severe salt-wasting syndrome to the less severe simple-virilizing form. The genotype-phenotype correlation is complex, and it is expected that the higher the number of mutations detected, the worse the phenotype would be. Moreover, when more than one mutation occurs in the same patient, the phenotype should be the result of the most severe mutation detected.
Case Presentation: This is the case of a 49-year-old Caucasian man with simple virilizing congenital adrenal hyperplasia, diagnosed at two years of age due to neonatal presentation of ambiguous genitalia. The karyotype was 46, XX, the phenotype was male, and hormonal evaluations highlighted cortisol deficiency, which required cortisone acetate replacement therapy. Gonads were removed during infancy, surgical interventions were performed to align physical attributes with his male gender identity, and the patient underwent testosterone replacement therapy. During adulthood, while compensation for cortisone acetate was reached, managing testosterone therapy proved to be challenging, achieved after the introduction of a low dose of dexamethasone (0.125 mg daily) and testosterone gel 2% 30 mg daily. Genetic analysis unraveled five different heterozygous pathogenic variants on the CYP21A2 gene. One mutation was detected on the maternal allele, while the remaining four were found on the second allele. Three of these mutations were proven to be pathognomonic for the salt-wasting form.
Conclusion: This case underscored the intricate and heterogeneous correlation between genotype and phenotype in simple-virilizing congenital adrenal hyperplasia, illustrated by a patient with a rare occurrence of five distinct pathogenic mutations on the CYP21A2 gene. Although three of the five mutations detected are related to the salt-wasting congenital adrenal hyperplasia, only a simple-virilizing form was detected here. Moreover, although it is supposed that the higher the number of mutations detected, the worse the phenotype should be, here we described a simple-virilizing form in a patient with five mutations on the CYP21A2 gene.
Introduction
Congenital adrenal hyperplasia (CAH) is an autosomal recessive disorder characterized by a defect in the enzymatic steroidogenic cascade, which impairs the physiological production of both cortisol and aldosterone. The most common enzyme altered in the pathogenesis of CAH, accounting for approximately 90-95% of all cases, is microsomal P450c21. This enzyme catalyzes the 21-hydroxylation of progesterone into deoxycorticosterone and of 17-hydroxyprogesterone (17-OHP) into 11-deoxycortisol, serving as precursors for mineralocorticoids and glucocorticoids, respectively1, 2. When this enzyme is deficient or ineffective, precursors such as 17-OHP accumulate, along with lesser amounts of progesterone, 17-hydroxypregnenolone, and androstenedione, leading to a varying degree of deficiency in the final compounds, cortisol, and aldosterone1, 3.
The overall prevalence of CAH is estimated to range from 1 in 10,000 to 1 in 20,000 live births worldwide, although geographical and ethnic variations exist. Based on the severity of enzyme dysfunctions, two recognized forms of CAH exist: classic and non-classic. Classic CAH is estimated to occur in approximately 1 in 15,000 to 1 in 16,000 live births worldwide4, 5 and can be further categorized into two clinical presentations: salt-wasting (SW) and simple virilizing (SV) forms4. SW CAH, representing 75% of classic cases, is associated with the most severe clinical manifestations due to the complete absence of 21-hydroxylase activity, resulting in total deficiency of cortisol and aldosterone production and thus a lack of hormones with mineralocorticoid activity. This phenotype is compounded by hyperandrogenemia, leading to the accumulation of precursors with androgenic activity, potentially causing sexual ambiguity in affected female newborns. In contrast, the SV form of CAH is characterized by reduced enzymatic activity, estimated to be less than 2%, resulting in varying degrees of hyperandrogenemia. This can lead to external genital virilization in female newborns and precocious puberty in males, without overt cortisol/aldosterone deficiency1, 6. Classic CAH may interfere with genital system development and is classified as a disorder of sexual differentiation (DSD)7. Alongside classic forms, non-classic forms are prevalent, affecting approximately 0.1 – 0.2% of the general Caucasian population (1). Non-classic CAH is less severe, as genetic mutations do not completely impair 21-hydroxylase activity, which remains preserved at 20-60% compared to physiological levels8. Symptoms of non-classic CAH are primarily associated with hyperandrogenism and are typically diagnosed during adolescence or young adulthood in females, who may experience oligomenorrhea, infertility, and hirsutism, while diagnosis in males can be challenging4, 9, 10. The therapeutic approach to CAH involves hormone replacement therapy, management of symptoms, and addressing potential complications11. The primary goals are to normalize hormone levels, manage electrolyte imbalances, and promote normal growth and development. Considering the high variability in the CAH phenotype, this treatment must be highly individualized and could involve hormone replacement therapy (with glucocorticoids and mineralocorticoids), androgen suppression, surgical interventions, and psychosocial support. This latter should be accurately evaluated since CAH can impact the quality of life substantially and in multifaceted ways12. Indeed, the diagnosis and management of CAH is well-known to affect physical health, psychological well-being, social interactions, and overall life satisfaction. Obviously, the extent of this impact varies depending on the severity of the condition, effectiveness of treatment, and individual coping mechanisms, but requires accurate evaluation from the clinician.
The 21-hydroxylase gene, CYP21A2, consists of 10 exons13 and is located on chromosome 6p21.34, 14. In the literature, several gene variations associated with CAH are described, including large deletions, gene conversions, insertions, and single-nucleotide variants9, 10, 15. The continual discovery of new CYP21A2 mutations has led to the identification of over 300 mutations to date13, 16. Although a strong correlation between genotype and phenotype is suggested, it is not universally confirmed13. The clinical presentation is typically heterogeneous and complicated by the occasional detection of more than one mutation on the same allele, occurring in 5-6% of cases17. Numerous mutations have been described in patients with CAH, and it is even possible for multiple mutations to coexist in the same patient, potentially leading to a more severe phenotype4. While case reports of two to three concomitant mutations are relatively common in the literature, finding more than three mutations in the same gene is rare4, 16.
Here, we present the case of a patient with five different heterozygous pathogenic variants in the CYP21A2 gene, resulting in an SV form of CAH.
Case Presentation
The Diagnosis and Childhood
A 49-year-old man presented at the Unit of Endocrinology in Modena, Italy, in 2022 with a previously established diagnosis of CAH. Inquiring about his medical history revealed that he was the only child of non-consanguineous healthy parents. During infancy, he attended medical consultations at the Pediatrics department in Modena due to ambiguous genitalia. At the age of two, a suspicion of SV CAH syndrome arose, as indicated by a 46, XX karyotype and the absence of signs or symptoms of the SW form during the perinatal period. Biochemical examinations revealed reduced cortisol serum levels, prompting the initiation of corticosteroid replacement therapy. The therapy was adjusted to maintain cortisol serum levels within reference ranges and prevent androgen excess. Throughout childhood, the patient was raised as male and underwent several surgical procedures for sex reassignment. Specifically, at the age of nine, he underwent partial removal of his vagina and complete removal of his uterus and ovaries at the University Hospital of Liege in Belgium. Subsequently, hypospadias correction surgery was performed, and testicular prostheses were implanted. Starting at the age of ten, replacement androgen therapy was added to the corticosteroid treatment to induce male pubertal development. During adolescence, the patient also underwent lower limb elongation surgery to address short stature. The patient's subjective gender identity has consistently been male.
| Date | Steroid therapy | Androgen therapy | ACTH (pg/mL) | 17-OHP (nmol/L) | Total Testosterone (nmol/L) |
| Reference ranges | 4.3-52 | 1.21-7.23 | 7.29 – 23.6 | ||
| July 2018 | Cortisone Acetate 25 mg daily | Testosterone gel 2%, 60 mg daily | 69 | 5.5 | >55.2 |
| November 2018 | Cortisone Acetate 25 mg daily | Testosterone gel 2%, 30 mg daily | 56 | n.a. | 1.7 |
| April 2019 | Cortisone Acetate 25 mg daily | Testosterone gel 2%, 30 mg daily | 75 | 11.4 | 5.2 |
| November 2019 | Cortisone Acetate 18.75 mg daily | Testosterone gel 2%, 30 mg daily | 151 | 62.0 | 53.0 |
| June 2020 | Cortisone Acetate 25 mg daily | Testosterone gel 2%, 20 mg daily | 171 | 52.0 | 12.1 |
| January 2021 | Cortisone Acetate 25 mg daily | Testosterone gel 2%, 20 mg daily | 187 | 76.3 | 33.9 |
| September 2021 | Cortisone Acetate 31.25 mg daily | Testosterone gel 2%, 20 mg daily | 157 | 152.6 | >55.2 |
| November 2021 | Cortisone Acetate 31.25 mg daily | Androgen withdrawal | 189 | 101.6 | 1.4 |
| January 2022 | Cortisone Acetate 31.25 mg + dexamethasone 0.125 mg daily | Testosterone gel 2%, 10 mg daily | 189 | 36.4 | >55.2 |
| May 2022 | Cortisone Acetate 25 mg + dexamethasone 0.187 daily | Testosterone gel 2%, 5 mg daily | n.a. | 8.0 | >55.2 |
| June 2022 | Cortisone Acetate 25 mg + dexamethasone 0.187 daily | Testosterone gel 2%, 5 mg daily | 36.4 | 8.3 | 0.3 |
| September 2022 | Cortisone Acetate 25 mg dexamethasone 0.187 daily | Testosterone gel 20 mg/g, 23 mg daily | 11.4 | 1.4 | 44.0 |
| December 2022 | Cortisone Acetate 25 mg + dexamethasone 0.187 mg daily | Testosterone gel 20 mg/g, 23 mg every other day | n.a. | n.a. | 1.0 |
| March 2023 | Cortisone Acetate 25 mg + dexamethasone 0.125 mg daily | Testosterone gel 20 mg/g, 11.5 mg daily | 20.2 | 3.5 | 23.2 |
| Mutation | Protein change | Site | Molecular Effect |
| c.293-13A/C>G | - | Intron 2 | Abnormal splicing of the CYP21A2 pre-mRNA, leading to: 1. Skipping of exon 3 during mRNA splicin 2. Retention of intron 2 within the mRNA transcript 3. Cryptic splice sites within intron 2 or exon 3, resulting in a transcript with altered sequence or premature termination codons |
| c.844G>T | p.Val282Leu | Exon 7 | Change in the DNA sequence of the CYP21A2 gene, affecting the structure and function of the enzyme |
| c.923dup | p.Leu308PhefsTer6 | Exon 7 | Lead to truncated protein is usually unstable and rapidly degraded within the cell |
| c.955C>T | p.Gln319Ter | Exon 8 | Affect the structure and function of enzyme, altering the enzyme's catalytic activity, substrate binding affinity, or stability |
| c.1069C>T | p.Arg357Trp | Exon 8 | Affect the structure and function of the enzyme, altering the enzyme's catalytic activity, substrate binding affinity, or stability. The specific consequences depend on the location of the amino acid change within the protein structure and its role in enzyme function. |


Genetic Examinations
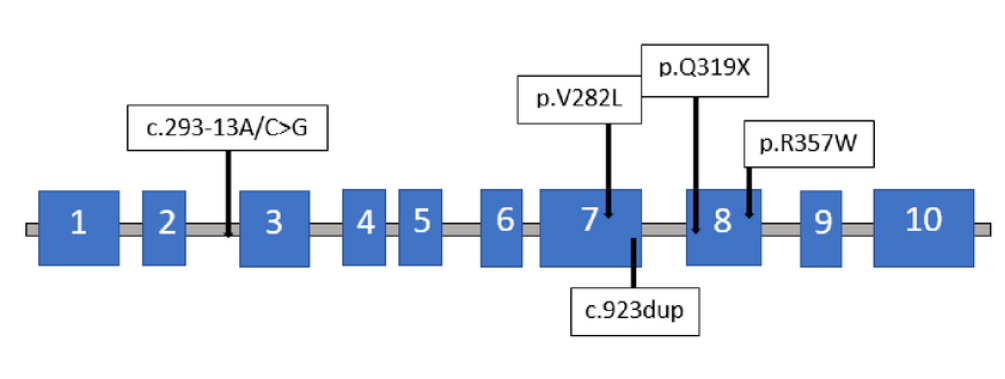
The genetic analysis of CYP21A2 was initially conducted when the patient was 34 years old at Hospital S. Orsola-Malpighi in Bologna, Italy, utilizing polymerase chain reaction (PCR) and direct sequencing of the entire gene, as previously described18. Five distinct pathogenic variants were identified on CYP21A2 (NM_000500.9) (Table 2): the c.293-13A/C>G (rs6467) variant was detected on the maternal allele (Figure 1), while the c.844G>T_p.Val282Leu (rs6471), c.923dup_p.Leu308PhefsTer6 (rs267606756), c.955C>T_p.Gln319Ter (rs7755898), and c.1069C>T_p.Arg357Trp (rs7769409) variants were found on the second allele. In November 2023, both the patient's and his mother's samples were investigated for the presence of deletion or duplication of the CYP21A2 gene using Multiplex Ligation-dependent Probe Amplification (MLPA). Two separate analyses were conducted: the first using the commercial kit (P050 lot D1-0222 from MRC-Holland, The Netherlands), and the second using a research-only kit containing specific probes for all the patient's variants (kindly provided by MRC-Holland, The Netherlands). The analysis revealed the presence of three copies of the gene. The maternal allele carried a copy of the CYP21A2 gene with the c.293-13A/C>G variant, while the paternal allele harbored two copies of the CYP21A2 gene: one with the p.Val282Leu and p.Leu308PhefsTer6 variants, and the other with the p.Val282Leu, p.Leu308PhefsTer6, p.Gln319Ter, and p.Arg357Trp variants. However, the lack of the father's sample precluded verification of the paternal allele configuration.
Adulthood
After discharge from the pediatric endocrinology service, the patient discontinued endocrinological follow-up until 2018 when he underwent an outpatient endocrinological examination at the Unit of Endocrinology in Reggio Emilia. Despite achieving clinical compensation of steroid replacement therapy with cortisone acetate, challenges with androgen replacement therapy were identified (Table 1). As expected, serum 17-OHP levels were consistently high, correlating directly with elevated adrenocorticotropic hormone (ACTH) serum levels. In 2022, a low dose of dexamethasone (0.125 mg daily) was added to steroid replacement therapy with cortisone acetate to reduce pituitary stimulation of the adrenal gland. This therapeutic adjustment resulted in a decrease in both ACTH and 17-OHP serum levels, reaching values within reference ranges (Table 1). Despite observing good clinical steroid compensation independent of ACTH/17-OHP serum levels and confirmed by normal blood pressure values, serum electrolytes, and blood glucose levels, total testosterone serum levels exhibited extreme variability (peak 116.15 nmol/L, nadir 1.73 nmol/L), which did not clearly align with the dose adjustments made despite reported optimal patient compliance (Table 1). Indeed, total testosterone serum levels seemed to be very sensitive to therapeutic changes, ranging from 116.15 nmol/L when the patient used Testosterone gel 2% 60 mg daily, to 1.73 nmol/L when the dose was reduced to 30 mg daily. Accordingly, when testosterone replacement therapy was suspended, the expected insufficient testosterone serum levels were confirmed (1.38 nmol/L). The detection of biochemical hypogonadism consistently coincided with signs such as decreased sexual desire and increased fatigue. Fluctuating testosterone levels persisted even after introducing dexamethasone and transitioning to another transdermal testosterone formulation, testosterone 20 mg/g gel (Table 1). To better understand the patient's hormonal imbalance, serum steroids were measured using the gold standard method, liquid chromatography/tandem mass spectrometry (LC-MS/MS) (Table 2). LC-MS/MS steroid levels were consistent with immunometric method measurements, confirming the absence of cortisol and aldosterone metabolites with an excess of their precursors. Finally, achieving testosterone serum levels within reference ranges was accomplished using 11.5 mg daily of testosterone gel 20 mg/g while maintaining corticosteroid therapy with both short and long-acting steroids. Consequently, symptoms related to hypogonadism improved with controlled hormonal levels. After one year of this new therapeutic approach, testosterone serum levels remained within reference ranges, with good compensation of the symptomatic picture.
During adulthood, the patient underwent multiple urological evaluations for episodes diagnosed as prostatitis. These evaluations included various radiological exams, ranging from trans-rectal ultrasound to magnetic resonance imaging (MRI) and abdominal computed tomography (CT) scans, revealing a prostate gland below normal volume limits. Additionally, a cystic pelvic formation measuring 5.0 x 3.3 x 4.0 cm with regular walls was identified, located between the bladder and the rectum, with a not definitively conclusive interpretation. A subsequent MRI conducted when the patient visited our Unit in 2023 confirmed the presence of a plausible prostate gland measuring 30 x 25 x 29 mm (Figure 2, Panel A), as well as an elongated and blind-bottomed formation with liquid content, measuring 33 x 32 x 56 mm, situated in the recto-vesical space (Figure 2, Panel B). This latter formation, consistent over time, was deemed compatible with either uterine or vaginal residue, with no discernible clinical consequences.
The patient provided informed consent for the publication of his clinical history.
Discussion
This case serves as a seminal illustration of the heterogeneous nature of CAH syndrome resulting from 21-hydroxylase deficiency. The extensive variability of the CYP21A2 locus is well-documented, with several genetic variants now recognized as pathogenic19. The phenotype is determined by the type of mutation detected and the number of mutations. Specifically, some CYP21A2 mutations are linked to the most severe phenotype, such as SW-CAH, while others are associated with milder forms. Although encountering multiple mutations within a single gene is common, cases with more than three mutations are rarely reported. It might be expected that a greater number of mutations would correlate with increased severity of the clinical presentation. However, the phenotype was moderate, characterized by an SV form, despite the identification of five different heterozygous mutations in the same individual, three of which are known to be associated with SW-CAH. Thus, in our case, the severity of the phenotype does not align with the detected genotype. The literature largely demonstrates that understanding genotype-phenotype relationships in CAH is crucial for accurate diagnosis, predicting disease severity, and tailoring treatment strategies to improve patient outcomes20, 21. Identifying multiple mutations responsible for CAH in a single patient allows for precise diagnosis, personalized treatment plans, and improved patient outcomes2, 22. This genetic information helps tailor glucocorticoid and mineralocorticoid dosing, predict enzyme activity, and manage adrenal crisis risks more effectively, optimizing hormone levels, reducing complications, and enhancing quality of life, including better psychological well-being and social integration. Additionally, genetic counseling for families becomes more accurate, aiding in carrier detection and informed family planning. Overall, detailed genetic insights contribute to the advancement of research and the development of targeted therapies.
The genotype of our patient is notably peculiar. Specifically, the first mutation detected in the maternal-origin allele, the CYP21A2 c.293-13A/C>G (rs6467), is a point mutation within intron 2, already described as causative for both SW and SV-CAH forms23. The correlation between this variant and the clinical phenotype remains incompletely understood. One possible mechanism is the synthesis of a small amount of correctly spliced product, hypothesized to activate a cryptic upstream three-splice acceptor site, causing aberrant splicing in an inconsistent manner and allowing for some correct splicing of mRNA capable of mitigating the severity of the disease10. The other four mutations detected in our patient are located on the second allele. The CYP21A2 c.844G>T p.Val282Leu mutation, resulting in an amino acid substitution in the I-helix region of the final protein chain, leads to steric collisions, reducing enzyme activity24, 25. This variant, supported by in vitro functional studies, is considered pathogenic for CAH 26. The CYP21A2 c.923dup p.Leu308PhefsTer6 mutation involves the insertion of a nucleotide, resulting in a premature termination codon and complete loss of 21-hydroxylase enzyme activity27. The CYP21A2 c.955C>T p.Gln319Ter mutation causes a nonsense mutation, resulting in an inactive protein3, 4. The CYP21A2 c.1069C>T p.Arg357Trp mutation describes a pathogenic variant associated with the SW phenotype28, 29, 30. Despite three of the five mutations being pathognomonic for SW-CAH, only an SV form was identified, indicating that the clinical manifestation is primarily influenced by residual enzymatic activity. Hence, given CAH resulting from 21-hydroxylase deficiency is a recessive disorder, the phenotype of each patient reflects the allele with the lesser degree of impairment1.
A total of 1,248 genetic variants causative for CAH are cataloged, with 460 affecting the translated region, and 52 mutations associated with classic CAH, primarily the SW form13. In our patient, three of these mutations were detected, although the phenotype did not manifest as severely as expected. It is probable that the combined effect of these mutations resulted in a different residual enzymatic activity, moderating the clinical presentation. Twenty-seven out of 35 associated mutations described in the literature are linked to SW-CAH, underscoring the potential additive effect of mutations on the clinical phenotype, which was not observed in our patient. Thus, our case report confirms that different phenotypes can be presented in patients with identical compound mutations24, 31, 32.
As a further complication, our patient experiences significant challenges in achieving testosterone serum levels within reference ranges during androgen replacement therapy. The absence of androgens renders testosterone serum levels nearly undetectable, as expected in the absence of testicles. Conversely, when low to intermediate doses of testosterone replacement therapy are administered, testosterone serum levels exceed the upper limit of reference ranges. This pattern persists across different testosterone formulations, suggesting that challenges in achieving adequate testosterone serum levels are not attributable to differences in transdermal absorption related to excipients. While the patient underwent a psychiatric evaluation in the past, which did not reveal any pathological features, further investigation into therapeutic compliance was not pursued. Therefore, we cannot definitively exclude non-compliance with androgen therapy, currently the most probable hypothesis.
The most intriguing aspect of the described case is the unique phenotype seemingly disconnected from the genotype. To the best of our knowledge, we are unable to justify this genotype/phenotype misalignment. Nonetheless, we could hypothesize that the presence of three copies of the CYP21A2 gene, instead of the typical two, could play a significant role in determining the phenotype.
Conclusion
This case presentation should serve as the starting point for future research aimed at advancing our understanding of genotype-phenotype relationships in CAH. Enhancing this understanding will facilitate more precise genetic counseling for CAH patients and improve the personalization of therapy. These efforts are essential for developing novel targeted therapies aimed at enhancing patient management and quality of life.
Abbreviations
ACTH - Adrenocorticotropic Hormone, CAH - Congenital Adrenal Hyperplasia, CT - Computed Tomography, CYP21A2 - Cytochrome P450 Family 21 Subfamily A Member 2 (a gene associated with CAH), DSD - Disorder of Sexual Differentiation, LC-MS/MS - Liquid Chromatography with Tandem Mass Spectrometry, MLPA - Multiplex Ligation-dependent Probe Amplification, MRI - Magnetic Resonance Imaging, PCR - Polymerase Chain Reaction, SV - Simple Virilizing, SW - Salt-Wasting, 17-OHP - 17-Hydroxyprogesterone
Acknowledgments
None.
Author’s contributions
GA, GS, SV, EM and DS performed the clinical evaluation of the patient. GA, GS and DS wrote the manuscript. SM performed genetic analyses. AF, ARM and MS revised the manuscript. All authors read and approved the final manuscript.
Funding
None.
Availability of data and materials
Data and materials used and/or analyzed during the current study are available from the corresponding author on reasonable request.
Ethics approval and consent to participate
Not applicable.
Consent for publication
Written informed consent was obtained from the patient for publication of this case report and any accompanying images. A copy of the written consent is available for review by the Editor-in-Chief of this journal.
Competing interests
The authors declare that they have no competing interests.
References
-
Speiser
P.W.,
Azziz
R.,
Baskin
L.S.,
Ghizzoni
L.,
Hensle
T.W.,
Merke
D.P.,
Endocrine Society
Congenital adrenal hyperplasia due to steroid 21-hydroxylase deficiency: an Endocrine Society clinical practice guideline. The Journal of Clinical Endocrinology and Metabolism.
2010;
95
(9)
:
4133-60
.
View Article PubMed Google Scholar -
El-Maouche
D.,
Arlt
W.,
Merke
D.P.,
Congenital adrenal hyperplasia. Lancet.
2017;
390
(10108)
:
2194-210
.
View Article PubMed Google Scholar -
White
P.C.,
Speiser
P.W.,
Congenital adrenal hyperplasia due to 21-hydroxylase deficiency. Endocrine Reviews.
2000;
21
(3)
:
245-91
.
PubMed Google Scholar -
Iezzi
M.L.,
Varriale
G.,
Zagaroli
L.,
Lasorella
S.,
Greco
M.,
Iapadre
G.,
A Case of Salt-Wasting Congenital Adrenal Hyperplasia with Triple Homozygous Mutation: review of Literature. Journal of Pediatric Genetics.
2021;
10
(1)
:
57-62
.
View Article PubMed Google Scholar -
Therrell
B.L.,
Padilla
C.D.,
Loeber
J.G.,
Kneisser
I.,
Saadallah
A.,
Borrajo
G.J.,
Current status of newborn screening worldwide: 2015. Seminars in Perinatology.
2015;
39
(3)
:
171-87
.
View Article PubMed Google Scholar -
Marino
R.,
Ramirez
P.,
Galeano
J.,
Perez Garrido
N.,
Rocco
C.,
Ciaccio
M.,
Steroid 21-hydroxylase gene mutational spectrum in 454 Argentinean patients: genotype-phenotype correlation in a large cohort of patients with congenital adrenal hyperplasia. Clinical Endocrinology.
2011;
75
(4)
:
427-35
.
View Article PubMed Google Scholar -
Lee
P.A.,
Mazur
T.,
Houk
C.P.,
DSD/intersex: historical context and current perspectives. Journal of Pediatric Endocrinology & Metabolism.
2023;
36
(3)
:
234-41
.
View Article PubMed Google Scholar -
Miller
W.L.,
Auchus
R.J.,
The molecular biology, biochemistry, and physiology of human steroidogenesis and its disorders. Endocrine Reviews.
2011;
32
(1)
:
81-151
.
View Article PubMed Google Scholar -
Kocova
M.,
Anastasovska
V.,
Falhammar
H.,
Clinical outcomes and characteristics of P30L mutations in congenital adrenal hyperplasia due to 21-hydroxylase deficiency. Endocrine.
2020;
69
(2)
:
262-77
.
View Article PubMed Google Scholar -
New
M.I.,
Extensive clinical experience: nonclassical 21-hydroxylase deficiency. The Journal of Clinical Endocrinology and Metabolism.
2006;
91
(11)
:
4205-14
.
View Article PubMed Google Scholar -
Abali
Z. Yavas,
Guran
T.,
Diagnosis and management of non-CAH 46,XX disorders/differences in sex development. Frontiers in Endocrinology (Lausanne).
2024;
15
:
1354759
.
View Article PubMed Google Scholar -
Balagamage
C.,
Arshad
A.,
Elhassan
Y.S.,
Said
W. Ben,
Krone
R.E.,
Gleeson
H.,
Management aspects of congenital adrenal hyperplasia during adolescence and transition to adult care. Clinical Endocrinology.
2023;
:
Online ahead of print
.
View Article PubMed Google Scholar -
Simonetti
L.,
Bruque
C.D.,
Fernández
C.S.,
Benavides-Mori
B.,
Delea
M.,
Kolomenski
J.E.,
CYP21A2 mutation update: comprehensive analysis of databases and published genetic variants. Human Mutation.
2018;
39
(1)
:
5-22
.
View Article PubMed Google Scholar -
Levine
L.S.,
Zachmann
M.,
New
M.I.,
Prader
A.,
Pollack
M.S.,
O'Neill
G.J.,
Genetic mapping of the 21-hydroxylase-deficiency gene within the HLA linkage group. The New England Journal of Medicine.
1978;
299
(17)
:
911-5
.
View Article PubMed Google Scholar -
Falhammar
H.,
Wedell
A.,
Nordenström
A.,
Biochemical and genetic diagnosis of 21-hydroxylase deficiency. Endocrine.
2015;
50
(2)
:
306-14
.
View Article PubMed Google Scholar -
Concolino
P.,
Costella
A.,
Congenital Adrenal Hyperplasia (CAH) due to 21-Hydroxylase Deficiency: A Comprehensive Focus on 233 Pathogenic Variants of CYP21A2 Gene. Molecular Diagnosis & Therapy.
2018;
22
(3)
:
261-80
.
View Article PubMed Google Scholar -
Shinagawa
T.,
Horikawa
R.,
Isojima
T.,
Naiki
Y.,
Tanaka
T.,
Katsumata
N.,
Nonclassic steroid 21-hydroxylase deficiency due to a homozygous V281L mutation in CYP21A2 detected by the neonatal mass-screening program in Japan. Endocrine Journal.
2007;
54
(6)
:
1021-5
.
View Article PubMed Google Scholar -
Barbaro
M.,
Lajic
S.,
Baldazzi
L.,
Balsamo
A.,
Pirazzoli
P.,
Cicognani
A.,
Functional analysis of two recurrent amino acid substitutions in the CYP21 gene from Italian patients with congenital adrenal hyperplasia. The Journal of Clinical Endocrinology and Metabolism.
2004;
89
(5)
:
2402-7
.
View Article PubMed Google Scholar -
Richards
S.,
Aziz
N.,
Bale
S.,
Bick
D.,
Das
S.,
Gastier-Foster
J.,
Laboratory Quality Assurance Committee
ACMG,
Standards and guidelines for the interpretation of sequence variants: a joint consensus recommendation of the American College of Medical Genetics and Genomics and the Association for Molecular Pathology. Genetics in Medicine.
2015;
17
(5)
:
405-24
.
View Article PubMed Google Scholar -
Hubska
J.,
K\kepczyńska-Nyk
A.,
Czady-Jurszewicz
K.,
Ambroziak
U.,
Characteristics of Congenital Adrenal Hyperplasia Diagnosed in Adulthood: A Literature Review and Case Series. Journal of Clinical Medicine.
2023;
12
(2)
:
653
.
View Article PubMed Google Scholar -
Hosomi
S.S.,
Salles
I.C.,
Bachega
T.A.,
Mutation distributions among patients with congenital adrenal hyperplasia from five regions of Brazil: a systematic review. Archives of Endocrinology and Metabolism.
2023;
67
(3)
:
427-41
.
View Article PubMed Google Scholar -
Merke
D.P.,
Auchus
R.J.,
Congenital Adrenal Hyperplasia Due to 21-Hydroxylase Deficiency. The New England Journal of Medicine.
2020;
383
(13)
:
1248-61
.
View Article PubMed Google Scholar -
Elmougy
F.,
Elsharkawy
M.,
Hafez
M.,
Atty
S.A.,
Baz
H.,
Ibrahim
A.,
Genetic profiling of CAH Egyptian children: rapid guide to clinical interpretation of common mutations. Journal of Endocrinological Investigation.
2021;
44
(1)
:
83-93
.
View Article PubMed Google Scholar -
New
M.I.,
Abraham
M.,
Gonzalez
B.,
Dumic
M.,
Razzaghy-Azar
M.,
Chitayat
D.,
Genotype-phenotype correlation in 1,507 families with congenital adrenal hyperplasia owing to 21-hydroxylase deficiency. Proceedings of the National Academy of Sciences of the United States of America.
2013;
110
(7)
:
2611-6
.
View Article PubMed Google Scholar -
Haider
S.,
Islam
B.,
D'Atri
V.,
Sgobba
M.,
Poojari
C.,
Sun
L.,
Structure-phenotype correlations of human CYP21A2 mutations in congenital adrenal hyperplasia. Proceedings of the National Academy of Sciences of the United States of America.
2013;
110
(7)
:
2605-10
.
View Article PubMed Google Scholar -
Tusie-Luna
M.T.,
Traktman
P.,
White
P.C.,
Determination of functional effects of mutations in the steroid 21-hydroxylase gene (CYP21) using recombinant vaccinia virus. The Journal of Biological Chemistry.
1990;
265
(34)
:
20916-22
.
View Article PubMed Google Scholar -
Narasimhan
M.L.,
Khattab
A.,
Genetics of congenital adrenal hyperplasia and genotype-phenotype correlation. Fertility and Sterility.
2019;
111
(1)
:
24-9
.
View Article PubMed Google Scholar -
Doleschall
M.,
Luczay
A.,
Koncz
K.,
Hadzsiev
K.,
Erhardt
É.,
Szilágyi
Á.,
A unique haplotype of RCCX copy number variation: from the clinics of congenital adrenal hyperplasia to evolutionary genetics. European Journal of Human Genetics.
2017;
25
(6)
:
702-10
.
View Article PubMed Google Scholar -
Chiou
S.H.,
Hu
M.C.,
Chung
B.C.,
A missense mutation at Ile172—-Asn or Arg356—-Trp causes steroid 21-hydroxylase deficiency. The Journal of Biological Chemistry.
1990;
265
(6)
:
3549-52
.
View Article PubMed Google Scholar -
Kor
Y.,
Zou
M.,
Al-Rijjal
R.A.,
Monies
D.,
Meyer
B.F.,
Shi
Y.,
Phenotype heterogeneity of congenital adrenal hyperplasia due to genetic mosaicism and concomitant nephrogenic diabetes insipidus in a sibling. BMC Medical Genetics.
2018;
19
(1)
:
115
.
View Article PubMed Google Scholar -
Xu
C.,
Jia
W.,
Cheng
X.,
Ying
H.,
Chen
J.,
Xu
J.,
Genotype-phenotype correlation study and mutational and hormonal analysis in a Chinese cohort with 21-hydroxylase deficiency. Molecular Genetics & Genomic Medicine.
2019;
7
(6)
:
e671
.
View Article PubMed Google Scholar -
Tang
P.,
Zhang
J.,
Peng
S.,
Wang
Y.,
Li
H.,
Wang
Z.,
Genotype-phenotype correlation in patients with 21-hydroxylase deficiency. Frontiers in Endocrinology (Lausanne).
2023;
14
:
1095719
.
View Article PubMed Google Scholar
Comments
Article Details
Volume & Issue : Vol 11 No 7 (2024)
Page No.: 6583-6591
Published on: 2024-07-31
Citations
Copyrights & License
This work is licensed under a Creative Commons Attribution 4.0 International License.
Search Panel
- HTML viewed - 1787 times
- PDF downloaded - 552 times
- XML downloaded - 103 times
