

Copyrights: Nurjannah Hanim Mohd Yassin, Mohd Rizal Mohd Zain, Intan Juliana Abd Hamid, Anis Munirah Mohd Kori, 2025. License: This work is licensed under a Creative Commons Attribution 4.0 International License.
Abstract
Background: Hemophagocytic lymphohistiocytosis is a syndrome characterized by excessive immune activation leading to severe systemic inflammation. It encompasses primary and secondary forms, with the latter often triggered by infections. Paediatric cases of melioidosis complicated by hemophagocytic lymphohistiocytosis are rare and diagnostically challenging.
Case presentation: We report the case of a 7-year-old girl who developed a fever for two months that was unresponsive to antibiotics. She was ultimately diagnosed with melioidosis-associated hemophagocytic lymphohistiocytosis after one month in the ward. She presented with multiorgan involvement, including bicytopenia, severe transaminitis, and neurological symptoms. Prompt initiation of immunoglobulin and steroids led to clinical improvement and resolution of the fever. Diagnostic confirmation relied on meeting HLH-2004 criteria despite limited genetic testing availability.
Conclusion: This case underscores the importance of early recognition and aggressive management in pediatric hemophagocytic lymphohistiocytosis secondary to melioidosis to mitigate potentially fatal outcomes.
Introduction
Hemophagocytic lymphohistiocytosis (HLH) is an excessive and ineffective inflammatory reaction caused by an inadequate immune system response, resulting in abnormal immune regulation. In HLH, T cells, natural killer (NK) cells, and macrophages are overactivated, leading to uncontrolled cytokine release. The term HLH refers to a variety of illnesses, including primary HLH (which includes familial HLH) and secondary HLH (infection-associated hemophagocytic syndrome and autoimmune-associated macrophage activation syndrome)1. Melioidosis is caused by Burkholderia pseudomallei, a bacterial species commonly found in soil and fresh surface water. It appears to be an opportunistic disease in adults that affects individuals with comorbidities and rarely results in death if diagnosed early and treated with specific antimicrobials. However, in children, mortality rates of up to 24% have been documented despite the absence of risk factors2. HLH is a rare complication of pediatric melioidosis that presents diagnostic challenges. The symptoms and signs are similar to those of other common pediatric illnesses. Case reports regarding the correlation between HLH and melioidosis in the pediatric population are scarce. We report on a 7-year-old girl who presented with pyrexia of unknown etiology, and the diagnosis of melioidosis complicating HLH was confirmed only after two months of fever. The patient responded well to intravenous immunoglobulin (IVIG) and steroids.
Case Report
A 7-year-old girl presented with a two-month history of prolonged fever. The documented temperature fluctuated between 39°C and 40°C. She was admitted to the district hospital and was administered intravenous (IV) antibiotics; initially, IV cefotaxime, which was subsequently upgraded to IV meropenem over a two-week period. Nevertheless, the fever did not subside. Several investigations were conducted during the admission (Table 1 ). The abnormal investigations revealed normochromic normocytic anaemia, transaminitis, and elevated C-reactive protein (CRP) and erythrocyte sedimentation rate (ESR). The other connective tissue disease (CTD), infection screening, blood and urine cultures were negative. Ultrasound of the abdomen showed no intra-abdominal abscess. Throughout the course of the fever, she had transient nonspecific myalgia and arthralgia, as well as cutaneous rashes that resolved spontaneously. She had no previous history of water sports, playing in the dirt and soil, or international travel. Otherwise, she had no constitutional symptoms, alopecia, malar rash, or arthralgia. The CRP and ESR were reducing in trend; meanwhile, her liver function test (LFT) was worsening while receiving IV antibiotics for a total duration of 14 days. The patient continued to have a high-grade fever with persistent normochromic normocytic anaemia. The parents requested discharge after two weeks of admission. They opted to obtain a second opinion at another hospital due to the extended hospital stay with an undefined diagnosis.
After being discharged from the district hospital for one week, she presented to our hospital. Upon her arrival at our centre, she was found to be febrile, with mild pallor and no signs of respiratory distress. Her vital signs were: pulse rate of 110 beats per minute, blood pressure of 90/65 mmHg, and temperature of 40°C. Neck examination revealed lymphadenopathy in the left cervical region, measuring 2 cm x 2 cm. Other systemic examinations were unremarkable. Repeated investigations revealed an increase in CRP, bicytopenia (anaemia and leukopenia), and a high ferritin level, as shown in Table 2. After consulting with an infectious disease specialist, she was administered IV trimethoprim/sulfamethoxazole due to the persistent fever and the increase in CRP. Several infection workups were conducted. Repeated blood and urine cultures were negative. Serial full blood pictures suggested infection, but no abnormal cells were seen. Echocardiography revealed normal findings with no evidence of vegetation in the heart. Ultrasound of the neck showed bilateral cervical lymphadenopathies, with the largest measuring 0.9 cm in short axis diameter in level V of the right cervical region. Fortunately, after eleven days of IV trimethoprim/sulfamethoxazole, the temperature, CRP, and ESR were decreasing in trend.
However, on the fourteenth day of her admission, she experienced abrupt episodes of status epilepticus, which were accompanied by the sudden onset of severe transaminitis, as illustrated in Table 3. Additionally, she developed septicemic shock necessitating inotropic support, and her temperature remained high. There were no space-occupying lesions on computed tomography (CT) of the brain. Lumbar puncture was not performed due to the parents' refusal to undergo the procedure. The ferritin level was significantly elevated, exceeding 100,000 ng/ml. The most probable diagnosis was HLH, considering a persistent fever with multiorgan involvement (bicytopenia, severe transaminitis, and brain involvement) and a significantly elevated ferritin level. The patient was initiated on IVIG 2g/kg and IV methylprednisolone due to her deteriorating condition and the potential for abrupt cardiorespiratory collapse. The antibiotic was escalated to intravenous meropenem. The melioidosis serology result came back positive with a titre of 1:80. One week after the initiation of IVIG and steroids, she was afebrile and her liver function was improving. The values of T-cells, B-cells, and natural killer cells (TBNK) were found to be low. She completed three weeks of IV meropenem and received IVIG and high-dose IV methylprednisolone after six weeks in the hospital. After one week of being afebrile in the ward, the patient was discharged. She was discharged with amoxicillin-clavulanic acid antibiotics for 12 weeks (to treat melioidosis) and two weeks of oral steroids. The patient was seen at the clinic for a follow-up appointment two weeks after discharge. She showed a good clinical manifestation, with her repeated TBNK cells normalized. She did not experience any side effects as a result of her prolonged use of steroids.
Discussion
The signs and symptoms of HLH are often nonspecific and can overlap with various common diseases, including infections, tumors, and rheumatological disorders. Therefore, HLH should be considered in the differential diagnosis for several clinical scenarios, such as: (1) fever of unknown origin, (2) hepatitis accompanied by coagulopathy (approximately 30% of HLH patients exhibit elevated transaminases over 100 U/L), (3) sepsis with multiple organ failure, and (4) lymphocytic encephalitis1. In this particular case, we faced a diagnostic challenge as the patient experienced persistent fever despite receiving broad-spectrum antibiotics. Prior to the onset of status epilepticus, there was a slight improvement in inflammatory markers, specifically CRP and ESR, which were trending downward. However, the patient subsequently developed severe transaminitis and status epilepticus, likely indicating hepatic encephalopathy. During this period, repeated serum ferritin levels were significantly elevated, alongside severe transaminitis and bicytopenia. The diagnosis of HLH was established using the HLH-2004 criteria, with the patient meeting 5 out of the 8 required criteria: 1. Fever, 2. Splenomegaly, 3. Cytopenias affecting at least two of three blood lineages (neutrophils < 1 × 109/L, hemoglobin < 9 g/dL, platelets < 100 × 109/L), 4. Serum triglycerides ≥ 3.0 mmol/L (≥ 265 mg/dL) or serum fibrinogen < 150 mg/dL, 5. Hemophagocytosis in bone marrow, spleen, or lymph nodes without malignancy, 6. Ferritin ≥ 500 mcg/L, 7. Soluble CD25 ≥ 2,400 U/mL (not available at our institution), 8. Reduced or absent NK cell activity or a genetic test confirming primary HLH3.
| 6/3/24 | 9/3/24 | 22/3/24 | |
|---|---|---|---|
| WBC ꝉ (10 9 /L) | 19.7 | 15.96 | 10.5 |
| Haemoglobin (g/dL) | 10.3 | 8.4 | 8.8 |
| Platelet (10 9 /L) | 365 | 488 | 426 |
| Sodium (mmol/L) | 136 | ||
| Potassium (mmol/L) | 3.6 | ||
| Urea (mmol/L) | 3.6 | ||
| Creatinine (µmol/L) | 29 | ||
| Calcium (mmol/L) | 2.26 | ||
| Phosphate (mmol/L) | 1.15 | ||
| ALP ‡ (U/L) | 191 | 209 | |
| ALT § (U/L) | 48 | 93 | |
| AST ¶ (U/L) | 103 | 207 | |
| Albumin (g/L) | 33 | 29 | |
| CRP ꝉ (mg/L) | 321.5 | 257.7 | |
| ESR ‡ (mm/60min) | 106 | 112 | 95 |
| BFMP § | No malaria isolated | ||
| Leptospirosis serology | Negative | ||
| Mycoplasma serology | Negative | ||
| Sputum AFB ¶ | No AFB seen | ||
| Sputum MTB C & S ꝉ | Negative | ||
| Rheumatoid factor | Negative | ||
| C3(g/L) | 1.97 | ||
| C4(g/L) | 0.30 | ||
| ANA ‡ | Negative | ||
| ASOT § | Negative |
| 1/4/24 (Day 1 admission) | 15/4/24 (Day 14 admission) | 22/4/24 | 1/5/24 (During follow up) | |
|---|---|---|---|---|
| WBCꝉ (10 9 /L) | 5.46 | 3.97 | 6.79 | |
| Haemoglobin (g/dL) | 10.3 | 6.9 | 10.7 | |
| Platelet (10 9 /L) | 365 | 86 | 155 | |
| Sodium (mmol/L) | 136 | 136 | 140 | |
| Potassium (mmol/L) | 3.8 | 4 | 3.8 | |
| Urea (mmol/L) | 2.7 | 8.2 | 1.9 | |
| Creatinine (µmol/L) | 34 | 68 | 21 | |
| Calcium (mmol/L) | 2.25 | |||
| Phosphate (mmol/L) | 1.58 | |||
| Lactate dehydrogenase (IU/L) | >2500 | |||
| ALP ‡ (U/L) | 218 | 703 | 411 | 150 |
| ALT § (U/L) | 19 | 336 | 168 | 22 |
| AST ¶ (U/L) | 58 | 4100 | 140 | 32 |
| Albumin (g/L) | 34 | 31 | 36 | 43 |
| CRP ꝉ (mg/L) | 201 | 54.7 | 5.4 | |
| ESR ‡ (mm/60 min) | >140 | 92 | 51 | |
| GGT § (U/L) | 228 | |||
| Triglycerides (mmol/L) | 1.84 | |||
| Cholesterol (mmol/L) | 3.78 | |||
| High-density lipoprotein (mmol/L) | 0.68 | |||
| Low-density lipoprotein (mmol/L) | 2.27 | |||
| Leptospirosis serology | Negative | |||
| Mycoplasma serology | Negative | |||
| Ferritin (ng/mL) | 3075 | >100000 | 15058 | 485 |
| Melioidosis serology | 1:80 | |||
| Rickettsial serology | Negative | |||
| EBV-IgM | Negative | |||
| ASOT | Negative | |||
| HBsAg | Non-reactive | |||
| HCV Ab | Non-reactive | |||
| HIV ELISA | Non-reactive | |||
| HSV 1 & 2 IgG | Non-Reactive | |||
| Toxoplasma IgM | Non-Reactive | |||
| CMV IgM | Non-Reactive | |||
| Rheumatoid factor | Negative | |||
| C3(g/L) | 2.23 | |||
| C4(g/L) | 0.26 | |||
| ANA ¶ | Negative | |||
| ASOT ꝉ | Negative | |||
| NK cell ‡ (cells/ μ L) | 71 | 812 | ||
| T cell (cells/ μ L) | 1106 | 2497 | ||
| B cell (cells/ μ L) | 185 | 356 |
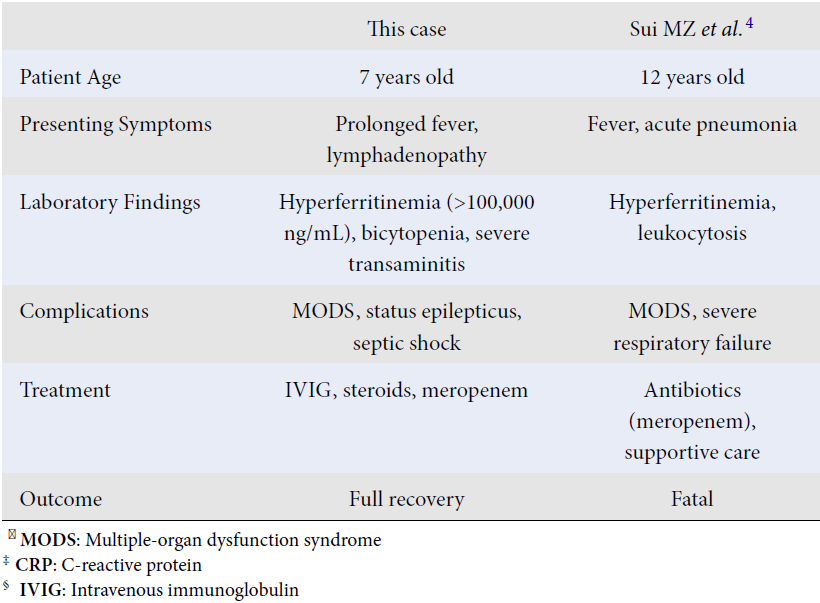
| This case | Sui MZ et al . 4 | Mohan A et al . 5 | |
|---|---|---|---|
| Patient Age | 7 years old | 12 years old | 6 years old |
| Presenting Symptoms | Prolonged fever, lymphadenopathy | Fever, acute pneumonia | Fever, respiratory distress |
| Laboratory Findings | Hyperferritinemia (>100,000 ng/mL), bicytopenia, severe transaminitis | Hyperferritinemia, leukocytosis | Hyperferritinemia, pancytopenia, elevated CRP ‡ |
| Complications | MODS ꝉ , status epilepticus, septic shock | MODS ꝉ , severe respiratory failure | MODS ꝉ , encephalopathy |
| Treatment | IVIG § , steroids, meropenem | Antibiotics (meropenem), supportive care | Antibiotics, steroids |
| Outcome | Full recovery | Fatal | Fatal |
Although the patient met 4 out of the 8 criteria, the diagnostic process was complicated by the fact that many of these tests are not routinely available in resource-limited settings. Nevertheless, the presence of cytopenia, significantly elevated ferritin, and liver function abnormalities are valuable indicators for distinguishing HLH from other clinical conditions in developing countries6. In this case, the refusal of lumbar puncture limited the ability to evaluate for central nervous system (CNS) involvement, a critical component in HLH diagnosis and management. This impacted diagnostic certainty by preventing confirmation of possible lymphocytic encephalitis or CNS infection, both of which could have strengthened the diagnosis of HLH. Alternative approaches included clinical assessment of neurological symptoms, such as status epilepticus, alongside biochemical markers like ferritin and transaminases, which strongly supported the diagnosis. Imaging studies, including a computed tomography (CT) brain scan, ruled out structural causes, further focusing the diagnosis on HLH. These steps compensated for the absence of lumbar puncture findings in guiding effective treatment. Genetic testing is not readily accessible at our institution and is only performed in specific cases deemed necessary by genetic specialists. It is likely that the patient experienced secondary HLH due to an infection, as indicated by positive serology for melioidosis. Additionally, NK cell activity normalized following treatment with IVIG and steroids, suggesting an acquired cause of HLH.
Pediatric melioidosis is reported to be less prevalent than in adults, accounting for 5 to 15% of all melioidosis cases4. The mechanisms and pathogenesis of severe melioidosis in children remain poorly understood. HLH has been observed in both children and adults without underlying conditions who have died from melioidosis, indicating a potential link to hyperferritinemic sepsis-induced multiple-organ dysfunction syndrome (MODS). The association between Burkholderia pseudomallei infection and HLH is linked to immune dysregulation caused by the pathogen’s ability to induce a cytokine storm through excessive release of pro-inflammatory cytokines like TNF-α, IL-6, and IFN-γ. This hyperinflammatory state can lead to the clinical features of HLH, including multiorgan dysfunction. Genetic susceptibility, such as variants in immune regulatory genes, may also play a role, though this is challenging to evaluate in resource-limited settings. Additionally, the overlap between hyperferritinemic sepsis-induced MODS and HLH suggests a shared pathophysiology2. The presence of hyperferritinemic MODS is a critical factor in severe cases of pediatric melioidosis, and early intervention may help reduce mortality and morbidity5. In this case, the patient suddenly developed severe transaminitis and status epilepticus alongside elevated ferritin levels. Given the rapid deterioration and biochemical changes, timely treatment was essential to prevent further decline. With the early initiation of IVIG and steroids, the patient responded positively, leading to a gradual improvement. Table 3 highlights the uncommon occurrence of full recovery in pediatric HLH cases caused by Burkholderia pseudomallei and underscores the critical role of prompt immunomodulatory treatment, alongside effective antibiotic therapy, in achieving positive outcomes, as demonstrated in this case.
In resource-limited settings, clinicians must rely on a combination of clinical judgment and the available HLH-2004 diagnostic criteria. Key laboratory markers such as hyperferritinemia, cytopenias, transaminitis, and elevated inflammatory markers (e.g., CRP, ESR) become essential substitutes for less accessible assays. Features like persistent fever, multiorgan dysfunction, and findings of hemophagocytosis in biopsy samples serve as practical diagnostic anchors. The presence of infection (e.g., Burkholderia pseudomallei in this case) can also help identify secondary HLH. Early empiric initiation of immunomodulatory therapy (e.g., IVIG, steroids) based on a presumptive HLH diagnosis is critical in settings where delayed testing may impact survival. Regular monitoring of clinical and biochemical response further supports decision-making in the absence of definitive testing. This case illustrates how combining clinical evaluation with limited but crucial lab findings can aid diagnosis and treatment even in resource-constrained environments.
Once HLH is suspected based on clinical and laboratory findings, a systematic treatment approach is initiated. The first step involves administering IVIG or a combination of IVIG and steroids. Continuous re-evaluation is crucial; if there is no improvement within 24 to 48 hours after starting steroids, chemotherapy following the HLH-04 protocol is introduced. Patients undergoing the complete HLH-04 protocol receive 8 weeks of chemotherapy, including cyclosporine and etoposide, alongside dexamethasone and IVIG. If patients show minimal or no clinical improvement and inflammatory markers remain elevated after 6 weeks of treatment, bone marrow transplantation (BMT) may be considered1. In this case, the patient's positive response to IVIG and steroids without the need for etoposide deviates from the HLH-04 protocol but was justified given the secondary HLH triggered by Burkholderia pseudomallei infection and the absence of genetic testing to confirm primary HLH. This approach minimized the risks associated with chemotherapy in a resource-limited setting while achieving clinical resolution. This case underscores the potential for tailored, less intensive therapy in specific secondary HLH cases, especially when prompt immunomodulatory treatment effectively stabilizes the patient, highlighting an area for further research in adapting HLH management to infection-associated cases.
Conclusions
HLH should be suspected in pediatric patients with prolonged fever unresponsive to antibiotics, particularly if accompanied by bicytopenia, hyperferritinemia, transaminitis, or neurological symptoms. In endemic areas, Burkholderia pseudomallei should be considered a potential trigger, especially in patients with environmental exposure. Early empiric immunomodulatory therapy with IVIG and steroids is crucial when HLH is strongly suspected, even before confirmatory tests are available, as it can significantly improve outcomes. Regular monitoring of clinical response and inflammatory markers is essential to guide treatment decisions.
Abbreviations
BMT: Bone marrow transplantation, CRP: C-reactive protein, CT: Computed tomography, CTD: Connective tissue disease, ESR: Erythrocyte sedimentation rate, HLH: Hemophagocytic lymphohistiocytosis, IG: Immunoglobulin, IV: Intravenous, LFT: Liver function test, MODS: Multiple-organ dysfunction syndrome, NK: Natural killer, TBNK: T and B natural killer cells
Acknowledgments
None.
Author’s contributions
AMMK, NHMY, MRMZ and IJAH participate in drafting the case report. AMMK is creating the outline of the case. All authors read and approved the final manuscript.
Funding
None.
Availability of data and materials
None.
Ethics approval and consent to participate
Not applicable.
Consent for publication
The parents of the patient gave written informed consent for the publication of this Case Report. A copy of the written consent is available for perusal by the Editor-in-Chief of this journal.
Competing interests
The authors declare that they have no competing interests.
References
-
Cleves
D.,
Lotero
V.,
Medina
D.,
Perez
P.M.,
Patiño
J.A.,
Torres-Canchala
L.,
Pediatric hemophagocytic lymphohistiocytosis: A rarely diagnosed entity in a developing country. BMC Pediatrics.
2021;
21
(1)
:
411
.
View Article PubMed Google Scholar -
Alvarez-Hernandez
G.,
Cruz-Loustaunau
D.,
Ibarra
J.A.,
Rascon-Alcantar
A.,
Contreras-Soto
J.,
Meza-Radilla
G.,
Description of two fatal cases of melioidosis in Mexican children with acute pneumonia: case report. BMC Infectious Diseases.
2021;
21
(1)
:
204
.
View Article PubMed Google Scholar -
Debaugnies
F.,
Mahadeb
B.,
Ferster
A.,
Meuleman
N.,
Rozen
L.,
Demulder
A.,
Performances of the H-score for diagnosis of hemophagocytic lymphohistiocytosis in adult and pediatric patients. American Journal of Clinical Pathology.
2016;
145
(6)
:
862-70
.
View Article PubMed Google Scholar -
Sui
M.Z.,
Wan
K.C.,
Chen
Y.L.,
Li
H.L.,
Wang
S.S.,
Chen
Z.F.,
Fatal hemophagocytic lymphohistiocytosis-induced multiorgan dysfunction secondary to Burkholderia pseudomallei sepsis: A case report. World Journal of Clinical Cases.
2023;
11
(30)
:
7372-9
.
View Article PubMed Google Scholar -
Mohan
A.,
Paranchothy
M.,
Segaran
S.,
Wong
R.S.,
Chor
Y.K.,
Podin
Y.,
Fatal Pediatric Melioidosis and the Role of Hyperferritinemic Sepsis-Induced Multiple-Organ Dysfunction Syndrome. The American Journal of Tropical Medicine and Hygiene.
2022;
107
(2)
:
393-6
.
View Article PubMed Google Scholar -
Smith
S.,
Munas
A.M.,
Hanson
J.,
Images in clinical tropical medicine hemophagocytic lymphohistiocytosis complicating melioidosis. American Journal of Tropical Medicine and Hygiene. The American Journal of Tropical Medicine and Hygiene.
2018;
99
(3)
:
557-8
.
View Article Google Scholar
Comments
Article Details
Volume & Issue : Vol 12 No 2 (2025)
Page No.: 7131-7137
Published on: 2025-02-28
Citations
Copyrights & License
This work is licensed under a Creative Commons Attribution 4.0 International License.
Search Panel
Pubmed
Google Scholar
Pubmed
Google Scholar
Pubmed
Google Scholar
Pubmed
Search for this article in:
Google Scholar
Researchgate
- HTML viewed - 1120 times
- PDF downloaded - 388 times
- XML downloaded - 89 times
